Синдром де Груши
Причина. Делеция короткого плеча 18-й хромосомы (возможны транслокационные и мозаические варианты).
Клиника. Низкая масса тела при рождении, профиль плоский или вогнутый. Маленький рост, микроцефалитная форма черепа, круглое лицо, высокое небо (иногда с расщелиной). Очаги облысения на голове либо тотальная алопеция. Характерна деформация зубов и ушных раковин, пупочечные и паховые грыжи. Аномалия кистей рук и пальцев, синдактилия пальцев ног. У мальчиков часто недоразвитие половых органов. Отмечается умственная отсталость.
Патогенез. У больных с грубой мозговой патологией резкое снижение продолжительности жизни. Косоглазие, мышечная гипотония, гипоплазия полового члена и мошонки у мальчиков и гипоплазия малых половых губ у девочек. Пороки сердца, иногда почек.
Диагностика. Исследование кариотипа, цитологическое обследование.
Лечение. Симптоматическое.
Частота. 1: 60000.
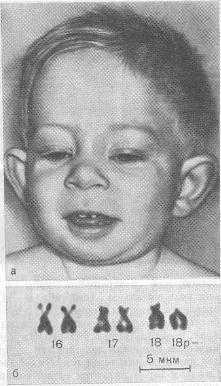
Синдром Лежена
Причина. Делеция длинного плеча 18-й хромосомы.
Клиника. Синдром мышечной гипотонии. Ребенок лежит на спине в позе «лягушки». Череп микроцефальной формы, уплощенное лицо с выступающим подбородком. Косоглазие, птоз, нистагм, снижение зрения, высокое твердое небо (иногда с расщелиной), своеобразная форма ушных раковин, нередко сужение слуховых органов. Интеллектуальное нарушение варьируется от легкой до олигофрении в степени идиотии.
Патогенез. Пороки сердца, иногда почек. Пороки развития зрительной системы.
Диагностика. Дерматоглифика (повышенное число завитков на пальцах рук), цитологическое обследование.
Частота. 1:60000.
Синдром Реторе
Причина. Частичная трисомия по короткому плечу хромосомы 9.
Клиника. Череп у новорожденных микробранхицефальный с уплощенным затылком. С возрастом брахицефалия уменьшается. Роднички широко открыты, имеется лобный шов. Характерны глазные аномалии: микро- или энофтальмия, страбизм, нарушение рефракции, эпикант, антимонголоидный разрез глаз, крупный нос с широким кончиком, опущены углы рта, короткая верхняя губа, «конские» зубы, дипластичное телосложение. Умственная отсталость варьируется от легкой до глубокой.
Патогенез. Глазные аномалии, у 25%- врожденные пороки сердца. Двигательные расстройства, нарушение координации.
Диагностика. Дерматоглифика, цитологическое обследование, кариологическое исследование.
Лечение. Отсутствует.
Синдром Брадера – Вилли
Причина. Утраивается участок 15-й хромосомы отцовского происхождения
Клиника. Мышечная гипотония, половое недоразвитие, ожирение, умственная отсталость. Низкая масса тела при рождении, пониженная температура. Потом развивается чрезвычайный аппетит. Деформированные низко расположенные ушные раковина и мягкие ушные хрящи, подковообразная форма рта, короткая губа, неправильный рост зубов. Диспропорциональность стоп и кистей. Нарушение осанки.
Патогенез. Частые грыжи, патология кистей и стоп. В пубертатном возрасте диабет. Взрослые страдают гиперсомнией, ишемической болезнью сердца, инфаркт миокарда.
Диагностика. Клиническое обследование.
Лечение. Симптоматическое.
studfiles.net
Синдром Ретта — причины, симптомы, диагностика и лечение
Синдром Ретта – генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Общие сведения
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Синдром Ретта
Причины синдрома Ретта
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы синдрома Ретта
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Третья стадия синдрома Ретта называется псевдостационарной, так как при ней мало заметны признаки прогрессирования заболевания. Обычно она длится от 4 до 15 лет, состояние больных стабильно, однако наблюдаются судорожные приступы, глубокая умственная отсталость, гиперкинезы. В большинстве случаев синдрома Ретта этот этап оканчивается в пубертатном периоде. Четвертая стадия синдрома Ретта характеризуется уменьшением частоты эпилептических припадков вплоть до их исчезновения при прогрессировании двигательных расстройств. Большинство больных полностью теряют подвижность, возникает атрофия мышц, сосудистые нарушения в нижних конечностях, что может привести к развитию трофических язв. Из-за слабости мышечного корсета спины при синдроме Ретта возникает сколиоз или другие формы искривления позвоночника.
Диагностика и лечение синдрома Ретта
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители. Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Прогноз и профилактика синдрома Ретта
Прогноз синдрома Ретта неблагоприятный, так как это заболевание неуклонно ведет к тяжелой умственной отсталости, а также к ряду двигательных и неврологических нарушений. Пациенты с этой патологией при соответствующем уходе и симптоматическом лечении способны доживать до 40-50 лет, однако риск внезапной смерти у них довольно высокий. Значительно ухудшает прогноз синдрома Ретта и снижает продолжительность жизни больных наличие пороков развития внутренних органов, что имеет место примерно в трети случаев. Основная причина летального исхода – дыхательная или полиорганная недостаточность и внезапная смерть, у взрослых больных также велик риск инсульта. Профилактика синдрома Ретта возможна только в виде пренатальной диагностики этого заболевания генетическими методами. При наличии дефектной формы гена MECP2 у мальчика нарушение формирования головного мозга и внутренних органов можно заметить при профилактических ультразвуковых исследованиях во время беременности.
www.krasotaimedicina.ru
причины, 18 симптомов, 4 этапа, 6 методов лечения
Истории детей с синдромом Ретта будто скопированы: беременность, роды проходят без осложнений, по шкале Апгар ребенку ставят высокий балл. Как это положено, в 3 месяца дети начинают держать голову. Сделать неуверенные шаги, сказать первые слова некоторые дети успевают, но в дальнейшем навыки теряются катастрофически быстро. За насколько недель ребенок теряет способность общаться с окружающими.
Что такое синдром Ретта
Синдром Ретта представляет собой прогрессирующее нейродегенеративное заболевание, которое влияет на развитие мозга ребенка и познавательные способности. Со временем он может вызвать серьезные проблемы с речью и коммуникацией, отсутствием координации и контроля мышц, непроизвольными движениями рук и замедлением роста.
Синдром был впервые открыт в 1966 году Андреасом Реттом, австрийским педиатром — неврологом. Синдром Ретта — редкое, но тяжелое расстройство головного мозга, которое поражает только одну из 12 000 девочек.
Синдром Ретта почти всегда поражает девочек, хотя мальчики могут заболеть в очень редких случаях. Младенцы, рожденные с синдромом Ретта, развиваются как обычные дети в течение первых нескольких месяцев их жизни. Они соответствуют типичным этапам развития, таким как визуальный контакт, взаимодействие с родителям, захватывание предметов. Однако, от 6 до 18 месяцев дети с синдромом Ретта начинают терять ранее приобретенные навыки, теряют интерес к игре и часто становятся все более раздражительными.
По мере роста у детей с синдромом Ретта начинают проявляться проблемы с коммуникацией и контролем движения мышц, что приводит к трудностям с движениями, таким, как ходьба. У них могут появляться проблемы с дыханием, кормлением и глотанием, а также у них могут быть судороги и нарушения сна. У большинства заболевших детей диагностируется интеллектуальная отсталость.

Современный подход к лечению синдрома Ретта фокусируется на симптоматическом лечении, улучшении движения и коммуникации, а также на поддержке пациентов и их семей. В настоящее время нет лекарств от этой болезни. Ученые всего мира занимаются изучением, исследованием патогенеза этой болезни, для того чтобы найти то самое заветное лекарство.
По распространённости в мире, это заболевание среди редких с подобной симптоматикой стоит сразу же после синдрома Дауна. В России по одним данным чуть больше сотни девочек с синдромом Ретта, по другим — 25 % от общего числа детей. Статистика хромает и вот ещё почему: синдром Ретта часто путают с детским церебральным параличом или аутизмом.Важно понимать, что хотя девочки и женщины, больные синдромом Ретта и испытывают трудности с общением, они имеют полный спектр эмоций и понимают больше, чем могут выразить.
Причины синдрома
Синдром вызван мутациями гена MECP2 на Х-хромосоме, одной из двух хромосом, определяющих пол человека. У девочек есть две Х-хромосомы, в то время как у мальчиков одна Х и одна У хромосома. Синдром Ретта чаще всего диагностируют у девочек, потому что у них есть вторая копия гена MECP2, которая способна работать правильно, и ребенок выживает.
Из-за отсутствия у мальчиков копии гена MECP2, при его мутации в единственной Х-хромосоме такие дети либо погибают внутриутробно, либо очень сильно больны. В большинстве случаев синдром Ретта не наследуется (не передается от родителя к ребенку). Вероятность болезни у последующих детей у родителей, у которых есть один больной ребенок, составляет приблизительно 1%.
До настоящего времени было идентифицировано более 200 мутаций гена MECP2, связанных с синдромом Ретта. При нормальном развитии человека ген MECP2 помогает создать белок (Methyl-CpG-связывающий белок 2), который регулирует активность других генов в организме. Когда уровень белка изменяется из-за дефектного гена, он заставляет другие гены в организме изменяться, что в конечном итоге влияет на нормальное развитие мозга.
Дети с расстройством могут проявлять широкий спектр симптомов. На это может влиять местоположение, тип и тяжесть мутации гена, могут быть и другие факторы. Необходимы дополнительные исследования, чтобы лучше понять ген MECP2 и как он влияет на различные функции организма.
Симптомы
Симптомы могут сильно варьироваться. Хотя генетическое изменение, которое вызывает синдром Ретта, присутствует до рождения, в большинстве случаев ребенок с синдромом Ретта будет расти и нормально развиваться в течение первых 6-18 месяцев жизни до появления симптомов.
Основные признаки
Симптомы синдрома Ретта на начальном этапе могут включать следующие проявления.
- Задержка развития.
- Потеря зрительного контакта.
- Отсутствие интереса к играм и общению.
- Замедление роста головы, приводящее к микроцефалии.
- Повышенная раздражительность.
- Снижение мышечного тонуса (гипотония).

По мере того, как заболевшие дети растут, они могут потерять навыки, которые они уже узнали, такие как речь, способность использовать свои руки для таких действий, как прием пищи. Дети с синдромом Ретта могут проявлять меньший интерес к людям и предметам, которыми они пользовались. Большинство детей с расстройством начинают проявлять классические движения синдрома Ретта в возрасте от 1 до 4 лет.
Они могут включать в себя сжатие, трение, скручивание, хлопание руками и повторяющиеся движения «рука в рот». По мере развития расстройства у детей будет все труднее контролировать свои мышцы, чтобы выполнять скоординированные двигательные движения. Это неврологическое состояние также называется апраксией или диспраксией.
Некоторые дети с синдромом Ретта могут ходить и поддерживать эту способность, другие могут в конечном итоге потерять это умение, в то время как некоторые могут никогда не ходить самостоятельно.
Дополнительные признаки
Дополнительные симптомы синдрома Ретта могут включать следующие признаки.
- Детская апраксия речи, в которой мозг ребенка испытывает трудности в координации движений мышц артикуляции аппарата, необходимых для формирования слогов и слов.
- Непроизвольные мышечные сокращения, вызывающие повторяющиеся движения.
- Мышечная слабость, суставные контрактуры и спастичность.
- Сколиоз.
- Симптомы, подобные тем, которые наблюдаются при болезни Паркинсона, такие как тремор, снижение мимики.
- Нарушения сна, в том числе трудности с засыпанием.
- Аномальное дыхание, которое включает в себя слишком быстрое или слишком медленное дыхание, дыхание с проглатывание воздуха.
- Проблемы с приемом пищи и глотанием.
- Желудочно-кишечные проблемы, включая рефлюкс и запор.
- Приступы панической атаки или беспокойства.
- Аутизм-подобные расстройства.
- Плохой рост и трудность в наборе веса.
После первоначальной регрессии, развитие имеет тенденцию стабилизироваться у большинства девочек. Некоторые навыки (отсутствие интереса к общению) могут улучшаться, в то время как другие ( двигательные навыки) могут оставаться такими же или постепенно ухудшаться. Другие симптомы, такие как аномалии дыхания или судороги, могут возникать и уходить со временем.
Несмотря на их многочисленные проблемы, у детей с синдромом Ретта есть своя уникальная личность, с симпатией и антипатией, они похожи на обычных детей.
Этапы болезни
- Начальный этап: от 6 до 18 месяцев. На этом этапе могут появиться первые симптомы синдрома. Появляется отсутствие интереса к игрушкам. Развитие двигательных навыков может быть замедленно. Рост окружности головы замедляется.
- Этап 2: от 1 до 4 лет. На этом этапе происходят самые большие изменения, часто быстрые, хотя и могут быть постепенными. Ребенок становится очень раздражительным. Повторяющиеся движения рук становятся очевидными, а целенаправленные движения рук теряются. Рост окружности головы замедляется. Могут возникнуть проблемы с дыханием.
- Этап 3: от 2 до 10 лет. На этом этапе быстрая регрессия второй стадии замедляется, и могут наблюдаться улучшения в поведении, уменьшается раздражительность. Контроль за движениями ухудшается. Потеря мышечного тонуса становиться выраженной. Могут начаться эпилептические припадки.
- Этап 4: 10+ лет. Этот этап характеризуется потерей движения. Теряется способность ходить. Может развиться сколиоз. Коммуникация и интеллект остаются на прежнем уровне. Повторяющиеся движения рук могут становиться меньше.
Диагностика синдрома Ретта
Существуют проблемы с ранней диагностикой, потому что не все врачи педиатры знакомы с первыми симптомами этого заболевания. Очень часто диагноз ставится поздно. Диагностика обычно начинается с выявления характерных симптомов болезни, а также сбора истории болезни и объективного осмотра.
Диагностика может включать следующие процедуры.
- Генетическое тестирование для выявления мутаций гена MECP2 для подтверждения клинического диагноза
- Неврологические исследования, такие как электроэнцефалограмма (ЭЭГ), которая оценивает электрическую активность в головном мозге и может обнаруживать наличие эпилептических приступов.
- Легочные функциональные тесты и тесты на сердечную функцию, включая электрокардиографию (ЭКГ), чтобы определить, насколько хорошо работают легкие и сердце ребенка.
В зависимости от симптомов, обнаруженных у ребенка, могут потребоваться дополнительные исследования для выявления любых проблем с приемом пищи и глотанием, желудочно-кишечных заболеваний или других медицинских проблем, которые могут возникнуть.
Лечение синдрома Ретта
В настоящее время нет никакого конкретного лечения синдрома Ретта. Лечение детей с расстройством является сложным и разнообразным и должно быть настроено для удовлетворения конкретных потребностей ребенка.
Лечение детей с синдромом Ретта часто включает следующие процедуры.
- Физиотерапия.
- Речевая терапия.
- Реабилитационная и поведенческая терапия.
- Специальная образовательная поддержка.
- Психосоциальная поддержка для ребенка и семьи.
- Питательная поддержка в виде добавок, специализированных диет. Назначается высококалорийная диета, которая помогает поддерживать достаточный вес, с использованием питательной трубки и других вспомогательных средств для кормления, если это необходимо.

Для лечения определенных симптомов, таких как судороги, назначается противоэпилептическая терапия.
Дети с синдромом Ретта также подвержены повышенному риску развития сколиоза и сердечной аритмии — оба проявления могут потребовать дополнительного лечения. Для контроля сердечного ритма назначаются бета-блокаторы или применяется кардиостимулятор.
Если сколиоз становится выраженным, используется корсет и спинальная хирургия, чтобы предотвратить дальнейшее развитие искривления позвоночника
Выяснилось, что терапевтическая верховая езда, плавание, дельфинотерапия, гидротерапия и музыкальная терапия благотворно влияют на течение болезни.
Важно постоянное выполнение физических нагрузок, занятия с логопедом, психологом, дефектологом. Если с ребенком не выполнять физических упражнений, не заниматься массажем, ЛФК, то он становиться лежачим и быстро теряет все приобретенные навыки.
Прогноз
Перспективы для детей с синдромом Ретта варьируют, и во многом зависят от прогрессирования и тяжести симптомов. Хотя дети с синдромом Ретта нуждаются в помощи в большинстве повседневных дел, многие могут приобретать некоторые самостоятельные навыки, такие как прием пищи или хождение в туалет. Хотя общение, как правило, ограничено, многие девочки могут научиться общаться другими способами, например, используя дополнительные устройства связи.
При оказании поддержки, люди с синдромом Ретта могут дожить до среднего возраста. Однако, из-за связанных с этим заболеванием состояний и осложнений для здоровья, люди с синдромом Ретта обычно имеют более короткую продолжительность жизни, чем в среднем у населения. Некоторые люди умирают в довольно молодом возрасте в результате осложнений, таких как аномалии сердечного ритма, пневмония и эпилепсия.
Продолжительность жизни во многом зависит от реабилитации. Ранняя реабилитация имеет потенциал к улучшению состояния.
kroha.info
Синдром Ротора: причины,проявление и лечение
Что такое синдром Ротора
Синдром Ротора – это редкая патология, которая носит врожденный характер и характеризуется умеренным пожелтением кожных покровов. Выявляется на первых неделях жизни младенца. Заболевание протекает легко, не вызывает неприятных ощущений и имеет весьма благоприятный прогноз.
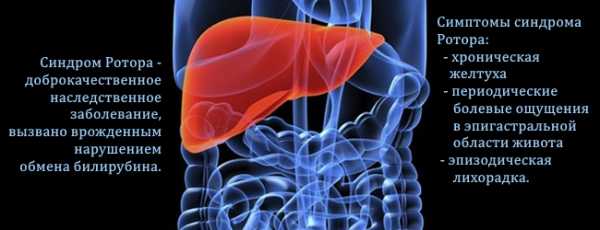
Подвержены патологии в одинаковой степени девочки и мальчики. Развивается болезнь в результате большого скопления билирубина в крови, которое возникает из-за нарушения работы печени, или в случае ее жировой дистрофии.
Причины развития заболевания
 Синдром Ротора – это наследственное заболевание, которое передается по аутосомно-доминантному типу. Так, при наличии у одного из родителей дефектного гена, все дети будут рождаться с данной патологией.
Синдром Ротора – это наследственное заболевание, которое передается по аутосомно-доминантному типу. Так, при наличии у одного из родителей дефектного гена, все дети будут рождаться с данной патологией.
Такая аномалия вызывает нарушение выведения билирубина в желчные протоки, в результате чего он в повышенном количестве скапливается в крови и окрашивает кожу в желтый цвет.
Симптомы синдрома Ротора
Основным признаком патологии является желтушность кожных покровов, что вызвано повышенным содержанием билирубина. При этом окрашиванию могут поддаваться и слизистые оболочки, а также склеры глаз. Желтуха может быть хронической – постоянное окрашивание кожи в желтый цвет и периодическая – пигментация наблюдается в период обострения и повышенной концентрации билирубина в крови.
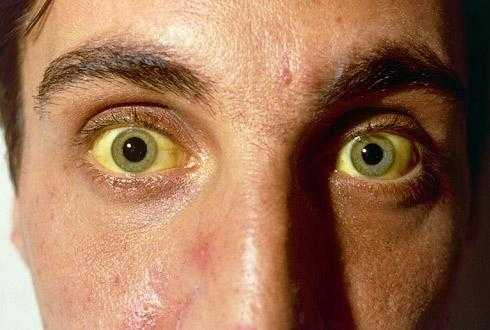
Порой у пациентов наблюдается потемнение мочи, может возникать чувство тяжести и боли в правом подреберье. Крайне редко больного донимают тошнота и рвота, горечь и неприятный привкус во рту, а также болезненные ощущения в животе. У пациента отсутствует аппетит, значительно снижается работоспособность и наблюдается быстрая утомляемость и постоянная слабость.
Диагностика болезни
Для диагностирования заболевания обязательно требуется консультация генетика и гастроэнтеролога. Важно установить наследственный и врожденный характер болезни. Это позволит сделать генетический анализ.
Для подтверждения диагноза выполняется биохимический анализ крови, который позволяет оценить уровень билирубина в крови. Общее исследование мочи проводится для выявление уробилиногена, который является продуктом восстановления билирубина.

Дополнительно выполняется инструментальная диагностика – ультразвуковое исследование печени и желчевыводящих путей, бромсульфалеиновая проба, холецистография с применением контраста. В некоторых случаях проводится биопсия печени с дальнейшим гистологическим исследованием образца тканей.
Лечение и профилактика синдрома Ротора
Поскольку синдром Ротора – это генетическое заболевание, оно не подлежит лечению. Возможно лишь устранение симптоматики. В частности, применяются желчегонные средства и кишечные сорбенты. Для выведения билирубина применяются специальные медикаментозные препараты. Также врач прописывает желчегонные средства и витаминно-минеральные комплексы.
В исключительных случаях, когда наблюдается чрезмерная концентрация билирубина, проводится переливание крови. Также рекомендовано проведение фототерапии. Такая процедура способствует расщеплению билирубина в крови на более простые соединения, которые проще выводятся из организма.
Важно обеспечить больному нормальные физические и эмоциональные условия, чтобы предотвратить рост билирубина. Так, стоит исключить сильные эмоциональные переживания, стресс, нервное перенапряжение и повышенные физические нагрузки.

Необходимо нормализовать питание – прием пищи должен быть частым, но в небольших количествах. Также важно обеспечить высококалорийный рацион. Из меню стоит исключить копчености, жирные продукты, консервацию, лук и чеснок, грибы, шоколад, приправы, газированные напитки. Важно следить и за потреблением соли. Суточная дозировка соли не должна превышать 10 г.
Вконтакте
Google+
24doctor.info
что это, причины, симптомы, диагностика, лечение
Синдром Ретта – врожденная патология головного мозга, характеризующаяся неполноценным развитием психики с нарушением интеллекта и приводящая к социальной дезадаптации. Это нервно-психическое генетическое заболевание обусловлено мутацией генов. Оно проявляется прогрессирующей умственной отсталостью преимущественно у девочек, гипотонусом мышц, шаткостью походки, сколиозом, запорами, утратой приобретенных навыков, дыхательными расстройствами, парезами и параличами. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой. Синдром у мальчиков встречается крайне редко. У них он несовместим с жизнью.
Синдром был открыт в 1954 году психоневрологом из Австрии А. Реттом, благодаря которому и получил свое название. В отдельную нозологию он был выделен намного позже: примерно через 10 лет. Причины и патогенез патологии в настоящее время остаются неизвестными. Ученые считают, что спонтанная мутация генов Х-хромосомы — источник данной проблемы.
В течение первого года жизни больной ребенок нормально развивается. Ближе к полутора годам у девочек с синдромом Ретта начинают пропадать приобретенные речевые, моторные и предметно-ролевые навыки. Их движения становятся стереотипными, однообразными и заторможенными, теряют целенаправленный характер. Они заламывают руки, сжимают пальцы, потирают кисти. Больные дети общаются с большим трудом: они часто погружаются в себя, замыкаются, потом резко кричат, беспричинно плачут. Неестественный смех быстро сменяется диким визгом. Дети с синдромом Ретта – необучаемые. Они не могут даже самостоятельно передвигаться.
Внешние признаки патологии – маленькие руки и ноги, замедление темпов роста головы, признаки микроцефалии. Болезнь неуклонно прогрессирует и приводит к выраженной умственной отсталости, потере навыков самообслуживания и обездвиженности. Пациенты с синдромом Ретта склонны к желудочно-кишечным расстройствам и эпиприпадкам.
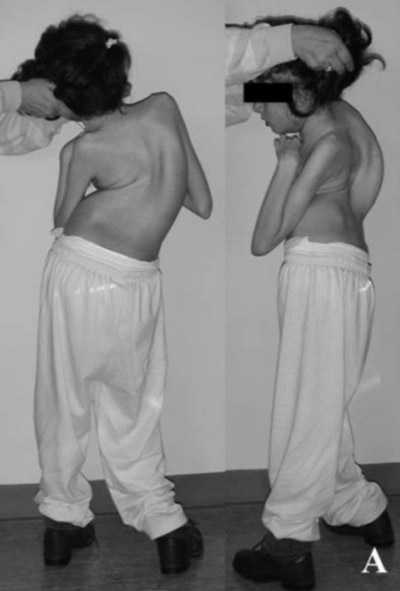
дети с синдромом Ретта
Диагностика заболевания заключается в проведении неврологического осмотра, МРТ головного мозга, ЭЭГ и молекулярно-генетического исследования. Синдром Ретта неизлечим. Больным показана симптоматическая терапия, целью которой является уменьшение выраженности отдельных проявлений недуга и облегчение общего состояния больного.
Причины
Этиопатогенетические особенности синдрома Ретта являются достаточно сложными. Они обусловлены взаимодействием различных генов и их влиянием на головной мозг. Первопричина патологии — спонтанная мутация гена Х-хромосомы, приводящая к прекращению его экспрессии. Этот ген кодирует специфический белок, регулирующий транскрипцию определенных участков ДНК. При синдроме Ретта нарушается ингибирование генов, что приводит к неправильному формированию нервной ткани. Еще одна теория происхождения патологии — ломкость одного из участков короткого плеча Х-хромосомы.
У больных нарушается ультраструктура нервных клеток: они уменьшаются в объеме, изменяется количество дендритов. Мозговые клетки не разрушаются, а начинают неправильно развиваться. Затрудненное образование нервной ткани никак не связано с процессом деструкции нейронов. Ученые отмечают, что при синдроме Ретта уменьшен размер головного мозга на 20% по сравнению с возрастной нормой. У пациентов преобладают процессы торможения в головном мозге. Это отражается на физиологии ЦНС, ее морфологическом строении и проявляется характерной клинической картиной.
Если в геноме данный ген отсутствует полностью, плод погибает внутриутробно. Летальное состояние в основном характерно для мальчиков. У плода женского пола имеется две хромосомы: одна нормальная, а вторая дефектная. Это позволяет дожить ему до рождения. У мужчин же имеется только одна X-хромосома. Если она несет дефектный ген, то у плода не остается нормальной копии гена, и он чаще всего погибает. В нетипичных случаях синдром Ретта развивается у мальчиков и сопровождается менее яркой клинической картиной.
В настоящее время к наследственной теории склоняется большинство ученых. Однако споры относительно этиопатогенетических факторов заболевания в ученом мире ведутся до сих пор.
Менее известная теория развития патологии — метаболическая. Смысл ее заключается в том, что недуг возникает в результате нарушения обмена веществ, вызванного дисфункцией митохондрий.
В настоящее время ни одна из этих теорий не доказана учеными. Они пока не смогли установить определенную закономерность, позволяющую утверждать стопроцентную взаимосвязь причины и следствия.
Факторы, способствующие развитию патологии:
- Неадекватное ведение беременности,
- Пристрастие будущей матери к вредным привычкам,
- Присутствие большого числа кровных связей в родословной человека.
Симптомы
Первые клинические признаки патологии возникают к концу первого года жизни. До года ребенок с синдромом Ретта растет и развивается обычными темпами.

Первые признаки недуга:
- Гипотермия,
- Гипергидроз,
- Бледность кожи,
- Трудности при переворачивании с живота на спину,
- Проблемы с ползанием и сидением.
Больные дети отстают от сверстников в росте, имеют непропорциональные руки и ноги по отношению к туловищу, поздно начинают ходить, часто срыгивают, быстро и поверхностно дышат, страдают эпизодами внезапной остановки дыхания и судорожными припадками, имеют задержку моторики и речи.
У больных детей возникают специфические движения рук, пропадают навыки удерживания предметов, появляются монотонные движения – перебирание пальцев или хлопки на уровне груди, сжимание кулачков, сплетение пальцев, поднесение рук к лицу, засовывание их в рот, покусывания рук или удары ими по разным частям тела. Движения рук полностью не исчезают, они становятся необычными. Ментальное развитие характеризуется умственной отсталостью и отсутствием познавательной деятельности, быстрой потерей приобретенных навыков. У больных развивается микроцефалия, появляются судорожные припадки, формируется сколиоз, обусловленный дистонией мышц спины. Непропорциональность головы по отношению к остальным частям тела становится очень заметной ближе к году.
Больные дети асоциальны. Они не реагируют на любые внешние раздражители и похожи своим поведением на больных аутизмом. Гипервозбудимость, приступообразный плач, абсолютная беспомощность — характерные признаки патологии. Дети с синдромом Ретта могут подолгу раскачиваться из стороны в сторону и переминаться с ноги на ногу. У некоторых отмечается высокой болевой порог. Они проявляют агрессию по отношению к себе и окружающим: бьют близких и себя, кусают ногти, царапаются.
К четырехлетнему возрасту рост мозга у больных детей останавливается полностью. Это приводит к дисфункции вегетативной нервной системы. У ребенка замедляется рост скелета, атрофируются мышцы, перестают нормально работать внутренние органы.
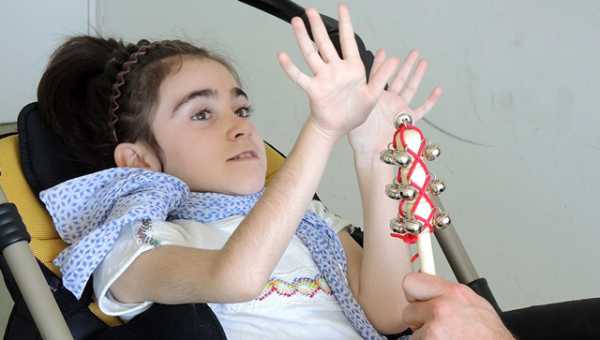
Эпиприпадки, бессонница и деменция с возрастом сменяются паркинсонизмом, стойкими нарушениями работы сердца и сосудов, потерей волос. У больных часто наблюдается дисморфизм лица, придающий им особый внешний вид. Женщины с синдромом Ретта фертильны, то есть способны к зачатию ребёнка.
Прогрессирование заболевания характеризуется определенной стадийностью:
- Первая стадия — стагнация. Она продолжается до 2,5 лет и проявляется следующими симптомами: снижением тонуса мышц, их слабостью, замедлением психомоторного развития, апатией, отсутствием интереса к окружающим людям и игрушкам, медленным ростом стоп и кистей в длину, недостаточным темпом роста головы, нарушением работы печени, сердца, ЖКТ. Лечение, начатое на таком этапе, может остановить прогрессирование синдрома Ретта.
- Вторая стадия — регресс длится в среднем два года. Клиницисты диагностируют эту стадию как энцефалит. Клинические признаки становятся более выраженными. Ребенок беспокойный, капризный, плохо спит и быстро утрачивает приобретенные навыки. На этой стадии нарушается процесс дыхания: периодически возникает апноэ, появляются признаки гипервентиляции – приступы учащенных и глубоких дыхательных движений. Дыхательные нарушения возникают во время бодрствования больного и исчезают во время сна. Характерны неврологические нарушения: атаксия, эпиприпадки, частые стереотипии. Целенаправленные движения сменяются хаотичными неконтролируемыми жестами. Судорожные припадки сопровождаются заламыванием рук с резким переходом от отрешенности и безэмоциональности к громкому крику. Симптоматическое лечение синдрома Ретта на этой стадии остается безрезультатным.
- Третья стадия относительной стабильности длится от 4 до 15 лет. Она отличается стабильным протеканием и возможным улучшением состояния ребенка. В это время признаки прогрессирования заболевания практически исчезают, нормализуется эмоциональный фон и сон. У больных учащаются судорожные приступы, формируется глубокая умственная отсталость, появляются гиперкинезы и экстрапирамидные расстройства, при которых подергивания сменяются «ступором» и онемением.
 Четвертая стадия – уменьшение частоты эпиприпадков вплоть до их полного исчезновения. Больные теряют подвижность, возникает атрофия мышц, сосудистые нарушения в ногах, трофические язвы, различные формы искривления позвоночника. Возрастание тонуса мышц в отдельных конечностях приводит к образованию контрактур. У больных снижается масса тела вплоть до крайней степени истощения. Несмотря на существенные отклонения в физическом развитии, у пациентов отмечается полноценное половое созревание. Последняя стадия наблюдается у взрослых и длится до конца жизни пациента.
Четвертая стадия – уменьшение частоты эпиприпадков вплоть до их полного исчезновения. Больные теряют подвижность, возникает атрофия мышц, сосудистые нарушения в ногах, трофические язвы, различные формы искривления позвоночника. Возрастание тонуса мышц в отдельных конечностях приводит к образованию контрактур. У больных снижается масса тела вплоть до крайней степени истощения. Несмотря на существенные отклонения в физическом развитии, у пациентов отмечается полноценное половое созревание. Последняя стадия наблюдается у взрослых и длится до конца жизни пациента.
 Четвертая стадия – уменьшение частоты эпиприпадков вплоть до их полного исчезновения. Больные теряют подвижность, возникает атрофия мышц, сосудистые нарушения в ногах, трофические язвы, различные формы искривления позвоночника. Возрастание тонуса мышц в отдельных конечностях приводит к образованию контрактур. У больных снижается масса тела вплоть до крайней степени истощения. Несмотря на существенные отклонения в физическом развитии, у пациентов отмечается полноценное половое созревание. Последняя стадия наблюдается у взрослых и длится до конца жизни пациента.
Четвертая стадия – уменьшение частоты эпиприпадков вплоть до их полного исчезновения. Больные теряют подвижность, возникает атрофия мышц, сосудистые нарушения в ногах, трофические язвы, различные формы искривления позвоночника. Возрастание тонуса мышц в отдельных конечностях приводит к образованию контрактур. У больных снижается масса тела вплоть до крайней степени истощения. Несмотря на существенные отклонения в физическом развитии, у пациентов отмечается полноценное половое созревание. Последняя стадия наблюдается у взрослых и длится до конца жизни пациента.Все вышеперечисленные симптомы позволяют определить, на какой именно стадии находится больной с синдромом Ретта. Признаки заболевания могут варьироваться в зависимости от скорости прогрессирования болезни и некоторых индивидуальных особенностей организма.
Диагностика
Диагностика патологии основывается на данных анамнеза жизни больного и его неврологического статуса, результатах ядерно-магнитного резонанса и электроэнцефалографии, а также молекулярно-генетического анализа.

Характерные признаки синдрома Ретта, позволяющие заподозрить данный недуг:
- нормальное развитие ребенка до 6-12 месяцев,
- мышечная гипотония,
- беспричинное беспокойство в раннем детстве,
- атаксия,
- частые эпиприпадки,
- утрата приобретенных навыков,
- умственная отсталость в более позднем возрасте,
- атрофия мышц,
- сколиоз,
- судороги.
Специалисты измеряют окружность головы ребенка и детально опрашивают родителей. Визуальный осмотр позволяет обнаружить отставание больного в росте от своих сверстников, уменьшение окружности головы, отсутствие речи.
Инструментальная диагностика:
- Результаты МРТ головного мозга — малый размер органа, сглаженность мозговых извилин, отсутствие четкой границы между серым и белым веществом.
- Данные энцефалографии – снижение фоновой активности головного мозга и резкое ослабление реакции на внешние раздражители.
- Медико-генетический анализ дает точную диагностическую информацию путем обнаружения мутации гена Х-хромосомы. Молекулярное кариотипирование позволяет обнаружить отклонения в гене, связанные с развитием заболевания.
- УЗИ внутренних органов — вспомогательный метод, позволяющий установить данную патологию. У больных детей обычно недоразвиты почки, сердце, печень.
- Рентгенография стоп и кистей – укорочение локтевой, четвертой плюсневой и пястной костей.
Лечение
Синдром Ретта — генетическое заболевание, которое абсолютно не поддается терапии. Современная медицина бессильна перед генетикой. Симптоматическое лечение, облегчающее жизнь больным, также сопряжено с рядом трудностей. Синдром Ретта в настоящее время лечат в специализированных реабилитационных центрах, которые имеются практически во всех крупных городах нашей страны. Специалисты центров проводят развивающие занятия, адаптируя маленьких пациентов к окружающему миру.

Медикаментозная терапия смягчает симптоматические проявления патологии и облегчает общее состояние. Больным назначают:
- Ноотропные препараты, улучшающие мозговую микроциркуляцию и стимулирующие работу органа – «Пирацетам», «Винпоцетин», «Цинноризин», «Мексидол», однако существенного эффекта от их применения не отмечено,
- Снотворные средства на основе мелатонина – «Мелаксен», «Циркадин», «Тразодон»,
- Антиконвульсанты, блокирующие приступы эпилепсии – «Карбамазепин», «Вальпарин»,
- Стимуляторы дофаминовых рецепторов – «Бромокриптин», «Толкапон», «Леводопа»,
- Психотропные препараты – «Фенибут», «Глицин», «Ноофен»,
- Препараты для лечения заболеваний сердца, печени, селезенки.
Лечебный рацион показан с целью предупреждения истощения больных. Им рекомендовано частое и дробное питание, употребление продуктов с высоким содержанием жиров, клетчатки и витаминов. Диету составляют индивидуально каждому больному для набора веса.
Физиотерапевтические процедуры, лечебный массаж, ЛФК — обязательные компоненты сложного лечебного процесса. Такие занятия способствуют развитию конечностей ребенка, повышают их гибкость, стимулируют мышечный тонус. Массаж и гимнастика поддерживают в исправном состоянии опорно-двигательный аппарат и противостоят дальнейшей деградации.
Лечение музыкой активно применяется при синдроме Ретта. Такая терапия благотворно воздействует на организм, успокаивает и стимулирует интерес к окружающему миру. Работа с дефектологом, психологом, остеопатом помогает ребенку улучшить навыки коммуникации и моторику. Остеопатия положительно влияет на состояние позвоночника. Мануальная терапия обычно сочетается с другими методами реабилитации. Хороший эффект дают занятия со специально обученными животными, гидрореабилитация, арт-терапия.
В настоящее время проводятся генетические и фармакологические исследования различных групп препаратов для лечения синдрома Ретта.
Прогнозы
Прогноз синдрома Ретта неоднозначный. Заболевание неуклонно ведет к тяжелой умственной отсталости, ряду двигательных и неврологических нарушений. Продолжительность жизни пациентов с этой патологией при соответствующем уходе и симптоматическом лечении в редких случаях составляет 40-50 лет. Но чаще всего больные погибают в раннем детском возрасте. Основная причина летального исхода – дыхательная или полиорганная недостаточность.
В настоящее время ученые разрабатывают специальные стволовые клетки, которых помогут побороть страшный недуг. Их вживление в организм позволит успешно противостоять прогрессированию болезни. Современные ученые экспериментально доказали, что синдром Ретта и его признаки вполне обратимы. Если восстановить функции гена, то можно устранить неврологические нарушения, которые провоцирует данная мутация. Эти исследования дают родителям и медикам большую надежду на восстановление большинства утраченных функций.

Поскольку синдром Ретта является генетическим заболеванием, предупредить его развитие невозможно. Профилактика патологии заключается в проведении ультразвукового скрининга всем беременным. Супружеские пары, имеющие в анамнезе хромосомные расстройства, с профилактической целью получают консультацию у генетика. Генетические тесты в процессе вынашивания ребенка позволяют выявить отклонение и принять решение о целесообразности прерывания беременности.
Считается, что нарушения генотипа человека провоцируют негативные факторы — сложная экологическая обстановка и вредные привычки. Специалисты дают общие рекомендации своим пациентам: вести здоровый образ жизни, правильно питаться, заниматься спортом и следить за своим самочувствием. Женщинам в период беременности также необходимо следить за адекватным протеканием гестации и регулярно посещать акушера-гинеколога.
Синдром Ретта – редкое заболевание, которое преимущественно диагностируется у девочек. Его невозможно победить окончательно. Современная медицина предлагает различные лекарственные препараты для уменьшения симптоматики недуга. Родителям не стоит отчаиваться, а следует помнить, что больной ребенок нуждается в бережном отношении, в любви и поддержке. Выполняя весь комплекс врачебных рекомендаций, можно значительно облегчить состояние малыша и улучшить его жизнь.
Видео: примеры детей с синдромом Ретта
sindrom.info
|
Поиск Лекций
В качестве примера синдрома со структурной перестройкой хромосом можно привести синдром делеции короткого плеча 5-й хромосомы — синдром 5р—, или синдром «кошачьего крика». Среди больных этим синдромом преобладают девочки. Частота встречаемости — 1:50 000 новорожденных. Характерные симптомы — это микроцефалия, круглое лицо (с возрастом оно вытягивается), широко расставленные глаза, антимонголоидный разрез глаз, эпикант, недоразвитие нижней челюсти, катаракта, косоглазие и другие глазные нарушения. Синдром получил свое название в связи с тем, что крик новорожденных с этим синдромом напоминает кошачье мяуканье. Выраженная умственная отсталость отмечается во всех случаях. Среди детей с глубокой умственной отсталостью на эту патологию приходится 1% из всех случаев синдрома. Трисомия-18 К синдромам с анеуплоидией относится трисомия-18, или синдром Эдвардса, встречающийся с частотой 1:7000. В данном случае лишней является хромосома 18-й пары. При этом синдроме преобладают девочки. Характерные симптомы синдрома — выступающий затылок, тонкие переносье и спинки 1, недоразвитие нижней челюсти, «птичий профиль», деформированные ушные раковины и тяжелые пороки развития внутренних органов (чаще всего сердца), в связи с чем такие дети умирают в раннем возрасте. До года доживают 10% детей с трсомией-18. Все больные отстают в умственном и физическом развитии. Известны (описаны) и другие аномалии хромосомы-18, включающие как структурные, так и геномные перестройки. Синдром де Груши Моносомия-18р. Частота встречаемости — 1: 60 000. Обычно ребенок рождается в срок, но с небольшой массой тела. В дальнейшем характерными признаками являются: маленький рост, микроцефальной формы череп, круглое лицо. Часто имеют место очаги облысения на голове либо тотальная алопеция. Характерны деформация зубов и ушных раковин, не редкость пупочных и паховых грыж на фоне мышечной гипотонии, аномалии кистей руки пальцев, синдактилия пальцев ног, «стопа-качалка». У мальчиков часто бывает недоразвитие половых органов, характерно резкое снижение продолжительности жизни у больных с грубой мозговой патологией. Реже наблюдаются более легкие формы интеллектуального дефекта с нормальной продолжительностью жизни. Характерно сочетание умственной отсталости с судорожным синдромом и различными речевыми расстройствами. Помимо делеции, возможны транслокационные и мозаичные варианты. Синдром Лежена Моносомия-18q, или синдром 18q — синдром Лежена. Делеция длинного плеча хромосомы-18. Встречается с частотой 1: 60 000. Девочки с этим синдромом рождаются в 1,5 раза чаще, чем мальчики. Одним из ранних характерных признаков считается синдром мышечной гипотонии: ребенок лежит на спине в «позе лягушки». Обращают на себя внимание микроцефальной формы череп, уплощенное лицо с выступающим подбородком. Характерна частота различных пороков развития зрительной системы, т.е. при данном синдроме имеет место дефект развития, включающий дефекты зрения и интеллекта. Среди нарушений зрительной функции преобладают колобомы, косоглазие, птоз, нистагм, снижение остроты зрения, атрофия зрительных нервов. Характерны своеобразная форма носа, рта, высокое твердое нёбо, иногда с расщелиной, своеобразная форма ушных раковин, нередко сужение или атрезия наружных слуховых проходов. Характерным признаком считается недоразвитие наружных половых органов, нередко отмечаются пороки сердца, почек. Интеллектуальные нарушения варьируются от легкой ^пограничной интеллектуальной недостаточности и даже нормального интеллекта) до олигофрении в степени идиотии. Синдром Патау К геномным мутациям относится также трисомия-13 — синдром Патау. Встречается синдром с частотой 1:6000 новорожденных. Дети рождаются с истинной пренатальной гипотрофией. В 50% случаев беременность осложняется многоводием. Типичным признаком является расщелина губы и неба. Как правило, дети страдают полидактилией. Характерны пороки развития органов зрения: катаракта, микрофтальмия, анофтальмия, циклопия. Череп неправильной формы, возможна тригоноцефалия, узкие глазные щели, запавшее переносье, деформированные низко расположенные ушные раковины, дефекты скальпа. Отмечаются рефлексорное сгибание костей, «стопа-качалка», пороки внутренних органов: врожденные пороки сердца, почек, желудочно-кишечного тракта, мочеполовой системы. Продолжительность жизни резко снижена <Ф5% детей погибает в возрасте до года). В развитых странах отмечается тенденция к увеличению продолжительности жизни таких больных: 15% детей доживают до пятилетнего возраста и 2—3% — до десятилетнего возраста. Во всех случаях отмечается выраженное психическое недоразвитие. Помимо трисомной (75%), встречаются транслокационная (20%) и мозаичная (5%) формы. Синдром Реторе С различными типами хромосомных аномалий может быть связана трисомия-9р — синдром Реторе. Предполагается, что в группе умственно отсталых детей трисомия по короткому плечу 9-й хромосомы занимает по частоте второе место после рома Дауна. Девочки с данным синдромом встречаются в два раза чаще, чем мальчики. Череп у новорожденных микробранхицефальный с уплощенным затылком. С возрастом брахицефалия уменьшается. Роднички широко открыты, имеется лобный шов. Характерны глазные аномалии: микро- или энофтальмия, страбизм, нарушения рефракции, гипертелоризм эпикант, антимонголоидный разрез глаз, крупный нос с широким кончиком, опущенные углы рта, короткая верхняя губа, «конские» зубы, диспластичное телосложение. У 25% детей врожденные пороки сердца. Умственная отсталость диагносцируется у всех больных. Выраженность интеллектуального дефекта варьируется от легкой до глубокой; типичны эмоциональная лабильность, повышенная психомоторная возбудимость, двигательные расстройства с нарушением координации движений. Среди цитогенетических вариантов синдрома Реторе — транслокационный, связанный с частичной трисомией (иногда с тетрасомией), «свободная» трисомия, мозаицизм предполагается, что характерные фенотипические проявления связаны с сегментом 9р21. Рекомендуемые страницы: Поиск по сайту |
|
poisk-ru.ru
Синдром Ретта — признаки генетической болезни у девочек, методы терапии и работа Ассоциации в России
Хотя это психоневрологическое заболевание является результатом наследственной патологии, первые месяцы жизни родители часто не подозревают о нездоровье своего малыша. Речь идет о разновидности дегенеративной врожденной болезни: синдром Ретта поражает нервную систему ребенка, останавливая его развитие, при этом клинические симптомы аналогичны с признаками прочих патологий, что осложняет своевременное диагностирование и лечение. Победить болезнь невозможно, но раннее распознавание возникновения синдрома дает шанс улучшить качество жизни.
Статьи по темеЧто такое синдром Ретта
В 1966 году невролог из Австрии Андреас Ретт подвел итоги многолетнего наблюдения за психическим регрессивном развитием 31 девочки. Регресс проявлялся как аутизм, потеря целенаправленных движений, сопровождался синдромом «сжимания рук». Исследования доктора не получили огласки в мире, лишь в 1983 году, благодаря статье шведского исследователя Бенгта Хагберга, болезнь выделили в отдельную нозологическую единицу, дав ей имя первооткрывателя Ретта. Заболевание встречается практически только у девочек с частотой 1:10000-1:15000.
Причины
Установлено, что болезнь Ретта – генетическое заболевание, которое может возникать как результат генетической мутации гена МЕСР2. Поврежденный ген блокирует своевременность определенных процессов мозга: его белок не выключает работу нескольких генов, после чего мозг ребенка теряет нормальный ход развития, останавливая рост на 4-м году жизни малыша.
Ген МеСР2 расположен в X-хромосоме. У женщин этих хромосом две, из которых при болезни Ретта у плода женского рода одна – нормальная, вторая – дефектная, за счет чего плод доживает до рождения. У мужчин такая X-хромосома одна, при наличии у нее аномального гена МЕСР2, плод ребенка без генной копии часто гибнет. По этой причине мальчики с болезнью Ретта рождаются редко. При том, что врожденная расположенность к заболеванию подтверждена, вопрос о природе этой патологии не закрыт.


Симптомы
При болезни Ретта у детей наблюдается характерная картина:
- замедляется общий рост;
- конечности и голова не развиваются, оставаясь маленькими;
- задерживается психическое, в том числе речевое и моторное развитие;
- при ходьбе не сгибаются колени;
- развивается сколиоз;
- появляются клинические судороги, приступы как при эпилепсии.
Практически у всех детей есть проблемы с дыханием:
- внезапная остановка – апноэ;
- срыгивание на фоне заглатывания воздуха – аэрофагия;
- быстрое частое поверхностное дыхание – гипервентиляция.
Характерный симптом – многократные частые движения, в основном руками: движение, похожее на мытье рук, потирание ушибленного места. Заболевший часто кусает стиснутые обслюнявленные кулачки. Выделяют 4 стадии с характерными клиническими особенностями:
- Возраст 4 месяца – 1,5-2 года. Рост головы и конечностей замедляется, мышцы слабеют, ребенок вялый, с полным отсутствием интереса к играм.
- Возраст 2-3 года. Теряются навыки ходить, говорить отдельные слова. Нарушается координация, начинаются движения руками, аномалии дыхания. Могут начаться судороги.
- Возраст 3-9 лет. Характеризуется умственной отсталостью в глубокой форме, эпилептическими припадками, двигательными нарушениями экстрапирамидных расстройств, когда подергивание тела сменяется резким общим ступором.
- Возраст от 10 лет. Наблюдается необратимость изменений вегетативной системы, двигательного аппарата, развивается кахексия (резкая потеря веса).


Диагностика синдрома Ретта
Своевременность и точность постановки диагноза осложнена тем, что первые шесть месяцев жизни малыша признаки болезни не проявляются, а в последующем не отличаются от детского аутизма. Помимо того, в перинатальный период развития плода расценивается как обычное и нормальное. Синдром Ретта у детей диагностируется по клинической картине и при первых подозрениях проводят аппаратное обследование: компьютерная томография, электроэнцефалограмма, УЗИ внутренних органов.
Первые признаки
Больные дети первые полгода выглядят совершенно здоровыми. Параметры тела, их общее развитие нормальны. Единственные подозрительные отклонения как признак синдрома – потливость ладошек, вялость мышц, бледная кожа, пониженная температура тела. К 4-5 месяцам уже становится заметно отставание в навыках движения, когда ребенок пытается переворачиваться, ползать.
Отличие синдрома Ретта от аутизма
| № п/п | Симптом | Признак проявления синдрома Ретта | Признак проявления аутизма |
| 1 | Отставание в развитии в возрасте от 6 месяцев до года | — | + |
| 2 | Движения рук | Однообразные и повторяющиеся движения рук в области пояса | Сложные разнообразные движения в любой зоне |
| 3 | Манипуляции с предметами | — | Постоянно повторяются, характерны для заболевания |
| 4 | Координация движений | Явные нарушения с переходом в полную обездвиженность | Походка и движения в норме, кажутся эмоциональными |
| 5 | Эпилептические припадки | Часто | Редко |
| 6 | Расстройства дыхания | Часто | — |
| 7 | Замедленный рост головы, кистей рук и стоп | Характерно для заболевания | — |
Лечение синдрома Ретта
На текущем этапе заболевание неизлечимо. Однако медикаментозным лечением в совокупности с реабилитационными методиками и диетой удается улучшить состояние ребенка, его качество жизни, предупредить деформацию тела. Для смягчения симптомов заболевания прописываются препараты противоэпилептические и противопаркинсонические, для улучшения мозговой функции, коррекции поведения, поддержки внутренних органов. Так как наблюдается быстрая потеря веса, больным требуется специальное питание с повышенным содержанием витаминов, жиров, клетчатки, калорий.
Прием лекарств для лечения патологии важно совмещать с реабилитационными методами:
- массаж, ЛФК;
- музыкотерапия:
- АВА-терапия;
- все виды терапии – занятий с животными;
- гидрореабилитация;
- арт-терапия;
- занятия с дефектологами, логопедами;
- лечение у мануальных терапевтов, остеопатов;
- игры, обучающие и развлекательные мероприятия.


Прогноз
Заболевания очень тяжелое, пока неизлечимое, но прогноз продолжительности жизни оптимистичен. Женщины с такой болезнью живут до сорока лет и дольше. Летальный исход наступает по причинам сильной эпилепсии, проблем с сердцем, перфорации желудка, дисфункции ствола головного мозга. Прогноз для мальчиков неутешителен: они не доживают до двух лет на фоне тяжелой энцефалопатии.
Ассоциация синдрома Ретта
В России для содействия исследованиям в области диагностики, реабилитации тяжелой болезни и для помощи семьям с больными людьми создана специальная Ассоциация. Отечественное и мировое медицинское научное сообщество активно ищет пути лечения генетической патологии, среди которых применение инсулина и стволовых клеток. Последние результаты демонстрируют возможность восстановления неврологических нарушений, что дает огромную надежду на восстановление моторных, мозговых, дыхательных функций людей при болезни Ретта.
Видео
 Синдром Ретта. Софья. Наш сайт Софья-ретта.рф
Синдром Ретта. Софья. Наш сайт Софья-ретта.рф
Внимание! Информация, представленная в статье, носит ознакомительный характер. Материалы статьи не призывают к самостоятельному лечению. Только квалифицированный врач может поставить диагноз и дать рекомендации по лечению, исходя из индивидуальных особенностей конкретного пациента.
Нашли в тексте ошибку? Выделите её, нажмите Ctrl + Enter и мы всё исправим!Рассказать друзьям:Статья обновлена: 13.05.2019
sovets.net