Мультифокальная моторная нейропатия с блоками проведения
Мультифокальная моторная нейропатия с блоками проведения была впервые описана R. Lewis, A. Sumner в 1982 г. Это — приобретенная аутоиммунная демиелинизирующая нейропатия, характеризующаяся развитием медленно прогрессирующей асимметричной слабости мышц конечностей, чаще дистальных отделов верхних конечностей, фасцикуляциями, крампи и отсутствием поражения сенсорных волокон периферических нервов. В дальнейшем было показано участие анти-ОМ1 ганглиозидов в формировании стойких иммунных комплексов с последующей фиксацией в миелиновой оболочке периферических нервов, нарушающих их целостность и препятствующих ремиелинизиации.
Заболевание наблюдается в 3—4 раза чаще у мужчин и проявляется в широком возрастном диапазоне (20—80 лет), чаще в возрасте 40—45 лет. Ключевое значение в диагностике имеет электронейромиография, основанная на регистрации и анализе биоэлектрических потенциалов мышц и периферических нервов. Электронейромиография позволяет обнаружить блоки проведения по двигательным волокнам при нормальном проведении по чувствительным.
Приводим описание соответствующего наблюдения.
Больная С., 27 лет, при поступлении в неврологическое отделение Республиканской клинической больницы предъявляла жалобы на слабость и похудение мышц в руках, больше левой, слабость в левой стопе.
Считает себя больной с 19-летнего возраста, когда появилась слабость в правой стопе, начала «пришлепывать». Через 2 года появилась легкая слабость в правой кисти; заметила, что «отошел мизинец». Еще через 2 года появились слабость и постепенное похудание мышц левой кисти и предплечья.
Неврологический статус: черепные нервы без патологии. Чувствительных расстройств нет. Сухожильные и периостальные рефлексы сохранены, с рук S≤D, коленные D=S, ахилловы рефлексы D≤S. Патологических рефлексов нет. Симптомов натяжения нет. Менингеальных знаков нет. В пробе Барре конечности удерживает. Координаторные пробы выполняет. В пробе Ромберга устойчива. Гипотрофия мышц левой кисти, слабость сгибателей и разгибателей левой кисти, гипотрофия мышц левого предплечья, слабость мышцы, отводящей мизинец правой кисти, слабость разгибателей правой стопы, гипотония икроножной мышцы справа.
Лабораторные показатели крови и мочи в пределах нормы, в том числе креатинфосфокинза 91 Е/л, общий белок 71 г/л. На рентгенограмме кистей выявлен остеопороз головок пястных костей.
При выполнении стимуляционной электронейромиографии были выявлены блоки проведения по правому локтевому нерву на уровне предплечья, левому локтевому — на предплечье, левому лучевому — в верхних третях предплечья и плеча, левому срединному — на уровне предплечья, правому большеберцовому — на голени (рис. 1). Проведение по сенсорным волокнам периферических нервов верхних и нижних конечностей было в норме.
1). Проведение по сенсорным волокнам периферических нервов верхних и нижних конечностей было в норме.
Рис. 1. Исследование левого локтевого нерва при отведении с мышцы, отводящей мизинец. Увеличение резидуальной латентности 3,93 мс (при норме 1,8 мс). Определяется блок проведения 2—3-й степени. Амплитуда М-ответа при стимуляции в точке «запястье» 3,34 мВ, в точке «локтевой сгтиб» 0,959 мВ (падение на 70—75%). Амплитуда сенсорного ответа 38,6 мкВ, скорость проведения 63,0 мс.
При выполнении игольчатой миографии в мышцах, иннервируемых пораженными нервами, определялись признаки выраженной нейрогенной перестройки. Отмечены незначительная спонтанная активность, единичные фасцикуляции, в полученных потенциалах двигательных единиц (ПДЕ) — резкое повышение амплитуды, сохранение нормальных значений длительности, выраженная полифазия в отдельных мышцах (рис. 2).
Рис. 2. Исследование локтевого сгибателя кисти слева. Средняя амплитуда ПДЕ 2051 мкВ (минимальная 693 мкВ, максимальная 3778 мкВ). Средняя длительность ПДЕ 11,6 мс (норма 11,0 мс). Выраженная полифазия ПДЕ.
Средняя длительность ПДЕ 11,6 мс (норма 11,0 мс). Выраженная полифазия ПДЕ.
На основании этих данных был поставлен диагноз «мультифокальная моторная нейропатия с блоками проведения».
Приведенное наблюдение подтверждает, что диагностика мультифокальной моторной нейропатии с блоками проведения вызывает достаточно большие трудности. Ключевую роль в постановке диагноза и дифференциальной диагностике играет электронейромиографическое исследование, которое позволяет выявить смешанное (аксонально-демиелинизирующее) поражение нерва с преобладанием демиелинизиации, неравномерность поражения сегментов нерва и асимметрию патологического процесса.
Дистальная моторная нейропатия, тип V (HMN5, дистальная спинальная амиотрофия), GARS м.
Исследуемый материал Цельная кровь (с ЭДТА)
Метод определенияСеквенирование.
Выдаётся заключение врача-генетика!
Исследование мутаций в гене GARS.
Тип наследования.
Аутосомно-доминантный.
Гены, ответственные за развитие заболевания.
GARS (GLYCYL-tRNA SYNTHETASE)
Ген расположен на хромосоме 7 в регионе 7p14.3, в том же самом регионе, что и ген наследственной моторно-сенсорной нейропатии тип 2D. Предполагается, что эти два заболевания могут быть аллельными. В пользу этого свидетельствует, также описание Sambuughim с соавт. в 1998 году семьи, в которой у части больных отмечались типичные клинические признаки наследственной моторно-сенсорной нейропатии 2Д типа, а у части — дистальной спинальной амиотрофии.
Определение заболевания.
Дистальная моторная нейропатия V типа – наследственное медленно прогрессирующее мышечное заболевание, вызываемое дегенерацией нейронов передних рогов спинного мозга. На сегодняшний день известны два гена, ответственных за развитие HMN5A: BSCL2 и GARS.
Патогенез и клиническая картина.
Ген GARS, кодируюет фермент глицил-тРНК-синтетазу. Мутации данного гена приводят к нарушению синтеза белков, необходимых для нормального проведения нервного импульса по аксонам. Первые признаки заболевания проявляются, как правило, в юношеском возрасте (16-17 лет), но начало заболевания может варьировать от 2 до 40 лет. Основными клиническими признаками являются слабость и гипотрофия мышц верхних конечностей, в особенности тенарной и межкостных мышц кисти. Заболевание прогрессирует медленно и в большинстве случаев ограничивается поражением дистальных отделов рук. У 40% больных отмечено распространение патологического процесса и на дистальные отделы ног. У 10% больных выявлено снижение вибрационной чувствительности в руках и наличие умеренно выраженной пирамидной симптоматики. При вовлечении нижних конечностей наблюдается слабость перонеальной мускулатуры, деформация стопы по типу pes cavus, потеря вибрационной чувствительности. Характерным признаком на электромиограме является отсутствие потенциала действия при наличии нормальных значений скоростей проведения импульса по периферическим нервам и дистальной латентности.
Мутации данного гена приводят к нарушению синтеза белков, необходимых для нормального проведения нервного импульса по аксонам. Первые признаки заболевания проявляются, как правило, в юношеском возрасте (16-17 лет), но начало заболевания может варьировать от 2 до 40 лет. Основными клиническими признаками являются слабость и гипотрофия мышц верхних конечностей, в особенности тенарной и межкостных мышц кисти. Заболевание прогрессирует медленно и в большинстве случаев ограничивается поражением дистальных отделов рук. У 40% больных отмечено распространение патологического процесса и на дистальные отделы ног. У 10% больных выявлено снижение вибрационной чувствительности в руках и наличие умеренно выраженной пирамидной симптоматики. При вовлечении нижних конечностей наблюдается слабость перонеальной мускулатуры, деформация стопы по типу pes cavus, потеря вибрационной чувствительности. Характерным признаком на электромиограме является отсутствие потенциала действия при наличии нормальных значений скоростей проведения импульса по периферическим нервам и дистальной латентности.
Частота встречаемости:
не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Antonellis, A., Ellsworth, R. E., Sambuughin, N., Puls, I., Abel, A., Lee-Lin, S.-Q., Jordanova, A., Kremensky, I., Christodoulou, K., Middleton, L. T., Sivakumar, K., Ionasescu, V., Funalot, B., Vance, J. M., Goldfarb, L. G., Fischbeck, K. H., Green, E. D. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 72: 1293-1299, 2003.
- OMIM
Мультифокальная моторная нейропатия Текст научной статьи по специальности «Клиническая медицина»
© ГОНЧАРОВА З. А., КОВАЛЕВА Н. С. УДК 616.83
001: 10.20333/2500136-2017-1-89-92
МУЛЬТИФОКАЛЬНАЯ МОТОРНАЯ НЕЙРОПАТИЯ
Гончарова З. А., Ковалева Н. С. Ростовский государственный медицинский университет, Ростов-на-Дону, 344022, Российская Федерация
А., Ковалева Н. С. Ростовский государственный медицинский университет, Ростов-на-Дону, 344022, Российская Федерация
P%’>м%. Мультифокальная моторная нейропатия относится к числу редких демиелинизирующих нейропатий. Данная лекция охватывает вопросы клиники, диагностики, дифференциальной диагностики и лечения указанной нозологии. В качестве примера, приводится собственное клиническое наблюдение случая мультифокальной моторной нейропатии.
Кл>ч%вы% слова: мультифокальная моторная нейропатия, блоки проведения, хроническая воспалительная демиелинизирующая полинейропатия.
Для цитирования: З.А. Гончарова, Н.С. Ковалева. Мультифокальная моторная нейропатия. Сибирское медицинское обозрение. 2017; (1): 89-92. В01:10.20333/2500136-2017-1-89-92
MULTIFOCAL MOTOR NEUROPATHY
Goncharova Z. A., Kovaleva N. S. The Rostov State Medical University of the Health Ministry of Russia, Rostov-on-Don
A., Kovaleva N. S. The Rostov State Medical University of the Health Ministry of Russia, Rostov-on-Don
Abstract. Multifocal motor neuropathy is a rare demyelinating neuropathies. This article covers the questions of clinic, diagnosis, differential diagnosis and treatment of this nosology. As an example, given their own clinical observation of case the multifocal motor neuropathy. Key words: multifocal motor neuropathy, holding blocks, chronic inflammatory demyelinating polyneuropathy. For citation: Z. A. Goncharova, N. S. Kovaleva. Multifocal motor neuropathy. Siberian Medical Review. 2017; (1): 89-92. DOI: 10.20333/2500136-2017-1-89-92
Актуальность
Мультифокальная моторная нейропатия (ММН) впервые была диагностирована и описана в 1980-х годах как один из вариантов хронической воспалительной демиелинизирующей полинейропатии (ХВДП) [4]. В дальнейшем в 1982 году R.A. Lewis и A.J. Sumner описывали это заболевание как редкую форму множественной мононевропатии при отсутствии тоннельного синдрома [2]. В 1986 году A. Pestronk et al. впервые предложили термин «мультифокальная моторная нейропатия» [16]. За счет трудностей диагностики и схожести клинических проявлений мультифокальной моторной нейропатии (ММН) с ХВДП и боковым амиотрофическим склерозом (БАС), данная нозология требует повышенного внимания, поскольку, в отличии от БАС, не является фатальным заболеванием и при своевременно начатой терапии не приводит к необратимой инвалидизации. В то же время тактика лечения таких больных кардинально отличается от таковой при ХВДП, что в свою очередь диктует необходимость повышенной настороженности при обследовании больных с подозрением на демиелинизирующие полинейропатии.
В дальнейшем в 1982 году R.A. Lewis и A.J. Sumner описывали это заболевание как редкую форму множественной мононевропатии при отсутствии тоннельного синдрома [2]. В 1986 году A. Pestronk et al. впервые предложили термин «мультифокальная моторная нейропатия» [16]. За счет трудностей диагностики и схожести клинических проявлений мультифокальной моторной нейропатии (ММН) с ХВДП и боковым амиотрофическим склерозом (БАС), данная нозология требует повышенного внимания, поскольку, в отличии от БАС, не является фатальным заболеванием и при своевременно начатой терапии не приводит к необратимой инвалидизации. В то же время тактика лечения таких больных кардинально отличается от таковой при ХВДП, что в свою очередь диктует необходимость повышенной настороженности при обследовании больных с подозрением на демиелинизирующие полинейропатии.
Эпидемиология Частота встречаемости ММН в популяции невелика и составляет 1-2 случая на 100 000 населения, чаще страдают мужчины, в среднем соотношение мужчины : женщины составляет 3 : 1. опк установила, что ММН ассоциирована с увеличением уровня антител класса М к ганглиозиду GM-1 в крови и отличается наличием ответа на терапию иммуномоду-ляторами [16]. По всей вероятности, вследствие этого при ММН имеется стойкая демиелинизация периферических нервов, так называемые блоки проведения возбуждения (БПВ) [7]. Стойко сохраняющаяся локальная демиелинизация вызывает вторичные аксональные нарушения и нейротрофические расстройства, клинически проявляющиеся в виде локальных мышечных аотрофий в зоне иннервации пораженных нервов [2,13].
опк установила, что ММН ассоциирована с увеличением уровня антител класса М к ганглиозиду GM-1 в крови и отличается наличием ответа на терапию иммуномоду-ляторами [16]. По всей вероятности, вследствие этого при ММН имеется стойкая демиелинизация периферических нервов, так называемые блоки проведения возбуждения (БПВ) [7]. Стойко сохраняющаяся локальная демиелинизация вызывает вторичные аксональные нарушения и нейротрофические расстройства, клинически проявляющиеся в виде локальных мышечных аотрофий в зоне иннервации пораженных нервов [2,13].
На сегодняшний день остается неясной причина селективного поражения двигательных волокон, не исключено, что так происходит вследствие разного антигенного состава двигательных и чувствительных волокон, либо их различной чувствительности к повреждающим факторам [4].
Клиническая картина
Заболевание начинается у большинства пациентов остро, однако, в ряде случаев возможно подострое развитие. Течение носит медленно либо скачкообразно прогрессирующий характер. Основное проявление ММН — формирование асимметричных парезов мышц конечностей, преимущественно в дистальных отделах, из-за поражения нескольких периферических нервов. Слабость в дистальных отделах рук встречается гораздо чаще, чем ног, и выявляется примерно у 95 % пациентов с ММН. Чаще всего страдают локтевой, срединный, лучевой, малоберцовый и большеберцовый нервы. Слабость разгибания отдельных пальцев является распространенным ранним симптомом [3]. Более чем у половины больных отмечаются фасцикуляции и крампи, из-за чего могут возникнуть ложные предположения о наличии у пациента бокового амиотрофического склероза (БАС). Сухожильные рефлексы меняются асимметрично, снижаясь либо выпадая с паретичных мышц и оставаясь не измененными с интактных.
Течение носит медленно либо скачкообразно прогрессирующий характер. Основное проявление ММН — формирование асимметричных парезов мышц конечностей, преимущественно в дистальных отделах, из-за поражения нескольких периферических нервов. Слабость в дистальных отделах рук встречается гораздо чаще, чем ног, и выявляется примерно у 95 % пациентов с ММН. Чаще всего страдают локтевой, срединный, лучевой, малоберцовый и большеберцовый нервы. Слабость разгибания отдельных пальцев является распространенным ранним симптомом [3]. Более чем у половины больных отмечаются фасцикуляции и крампи, из-за чего могут возникнуть ложные предположения о наличии у пациента бокового амиотрофического склероза (БАС). Сухожильные рефлексы меняются асимметрично, снижаясь либо выпадая с паретичных мышц и оставаясь не измененными с интактных.
Чувствительные расстройства при ММН отсутствуют, однако у некоторых больных могут быть жалобы на онемение либо парестезии. Атрофии мышц формируются достаточно медленно, по мере развития вторичной аксональной дегенерации. Однако, иногда атрофия может развиваться достаточно быстро (еще до развития значимого аксонального повреждения), что связывают с нарушением нейротрофического обмена между нервом и мышцей из-за накопления в синаптической щели аутоантител [4]. Поражение вегетативной нервной системы, черепных нервов, нервов иннер-вирующих дыхательную мускулатуру не характерно. Степень инвалидизации в целом коррелирует с продолжительностью заболевания [8,12]. Рабочая группа экспертов разработала клинические критерии диагностики ММН [3,5]. К ключевым критериям относятся медленно или скачкообразно прогрессирующая локальная асимметричная слабость в конечностях, сохраняющаяся более месяца. Двигательные расстройства выявляются не менее чем в двух двигательных нервах. Если симптомы распространяются только на 1 двигательный нерв, то ставится вероятный диагноз. Незначительные чувствительные расстройства в дебюте заболевания (но могут развиваться с течением времени).
Однако, иногда атрофия может развиваться достаточно быстро (еще до развития значимого аксонального повреждения), что связывают с нарушением нейротрофического обмена между нервом и мышцей из-за накопления в синаптической щели аутоантител [4]. Поражение вегетативной нервной системы, черепных нервов, нервов иннер-вирующих дыхательную мускулатуру не характерно. Степень инвалидизации в целом коррелирует с продолжительностью заболевания [8,12]. Рабочая группа экспертов разработала клинические критерии диагностики ММН [3,5]. К ключевым критериям относятся медленно или скачкообразно прогрессирующая локальная асимметричная слабость в конечностях, сохраняющаяся более месяца. Двигательные расстройства выявляются не менее чем в двух двигательных нервах. Если симптомы распространяются только на 1 двигательный нерв, то ставится вероятный диагноз. Незначительные чувствительные расстройства в дебюте заболевания (но могут развиваться с течением времени).
Критериями исключения являются признаки поражения верхнего мотонейрона, бульбарные расстройства, чувствительные нарушения более явные, чем незначительное снижение вибрационной чувствительности на ногах и диффузная симметричная слабость в течение первых недель от начала заболевания Диагностика
Золотым стандартом в диагностике ММН является электро-нейромиография (ЭНМГ), в частности стимуляционная, обнаруживающая множественные парциальные блоки проведения по двигательным волокнам (моторные блоки проведения — МБП) вне мест типичной компрессии, при нормальном проведении по сенсорным волокнам. Для максимально эффективного выявления блоков проведения используется методика пошагового исследования нерва предложенная Л. Ютига. Для более точного определения пораженного участка, шаг исследования нерва вместо обычных 10 см составляет 1-2 см [2,17]. Из-за наличия блока проведения развивается снижение амплитуды либо площади М-ответа, полученного при стимуляции проксимального отдела нерва, относительного такового при более дистальной его стимуляции [4]. М. Гехт с соавт. выделяют блоки проведения 1-й степени — снижение площади последующего М-ответа по отношению к предыдущему в диапазоне от 25 до 49 % при увеличении длительности М-ответа не более 15 % и блоки проведения 2-й степени — снижение площади последующего М-ответа по отношению к предыдущему на 50 % и более по отношению к предыдущему ответу при увеличении длительности М-ответа не более 15 % [2,3]. Также на ЭНМГ могут быть выявлены и другие признаки демиелинизации (увеличение терминальной латентности, падение амплитуд и дисперсия моторных ответов).
Для максимально эффективного выявления блоков проведения используется методика пошагового исследования нерва предложенная Л. Ютига. Для более точного определения пораженного участка, шаг исследования нерва вместо обычных 10 см составляет 1-2 см [2,17]. Из-за наличия блока проведения развивается снижение амплитуды либо площади М-ответа, полученного при стимуляции проксимального отдела нерва, относительного такового при более дистальной его стимуляции [4]. М. Гехт с соавт. выделяют блоки проведения 1-й степени — снижение площади последующего М-ответа по отношению к предыдущему в диапазоне от 25 до 49 % при увеличении длительности М-ответа не более 15 % и блоки проведения 2-й степени — снижение площади последующего М-ответа по отношению к предыдущему на 50 % и более по отношению к предыдущему ответу при увеличении длительности М-ответа не более 15 % [2,3]. Также на ЭНМГ могут быть выявлены и другие признаки демиелинизации (увеличение терминальной латентности, падение амплитуд и дисперсия моторных ответов). М1, GM1 и GM2, но могут быть и моноспецифичными [6,10,15,18].
М1, GM1 и GM2, но могут быть и моноспецифичными [6,10,15,18].
По данным исследования, проведенного в Первом ММГУ им. И.М. Сеченова на 7 больных с достоверным диагнозом ММН, чувствительность определения анти-GM1 при данном заболевании составила 71,4%, специфичность — 90%. Вероятность заболевания при положительном результате теста составляет 38,4 %, при отрицательном — 2,7% [6,10].
Учитывая, что, согласно данным литературы, частота вы-являемости анти-GM1 IgM при ММН варьирует от 30 до 80 %, отрицательные результаты иммунологического исследования при наличии других критериев данный диагноз не исключают. Таким образом, в настоящее время обнаружение повышения уровня анти-GM1-аутоантител является вспомогательным диагностическим критерием при данном заболевании, оказывающим помощь в постановке окончательного диагноза в сложных случаях. Эти антитела, однако, не являются диагностическим маркером для ММН, так как они могут определяться у больных с другими иммуноопосредованными нейропатиями [6,1].
Дифференциальная диагностика
Дифференциальная диагностика ММН заслуживает особого внимания, поскольку за счет схожести клинических проявлений, у этой когорты больных в ряде случаев, ошибочно диагностируется БАС, что может сопровождаться отказом от активной терапии и, соответственно, привести к инвалидизации больного, кроме того, эти пациенты живут с постоянным чувством страха неизбежной и скорой смерти. В то время, как мультифокальная моторная поллинейропатия является заболеванием поддающимся лечению с использованием иммуномодуляторов. Следует помнить, что при БАС слабость в мышцах не ограничивается зонами иннервации отдельных нервов, поражение носит более симметричный характер, амиотрофии нарастают агрессивнее и формируются быстрее, а также отсутствуют четкие блоки проведения при электрофизиологических исследованиях. При наличии у больного бульбарных расстройств и четких пирамидных нарушений диагноз БАС не вызывает сомнений.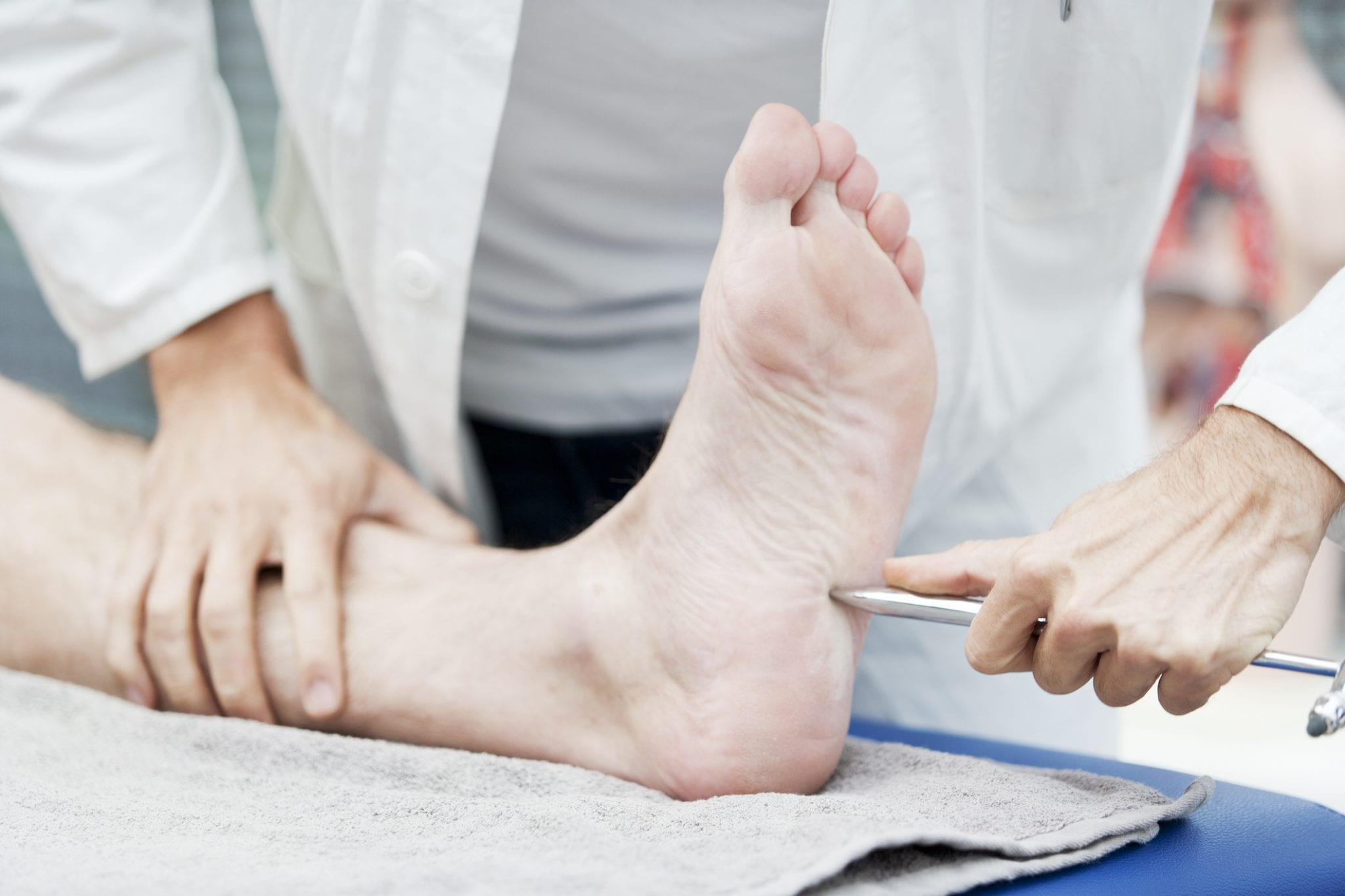
В ряде случаев больным с ММН устанавливается диагноз хронической воспалительной демиелинизирующей полиней-ропатией (ХВДП). Для ХВДП характерно более симметричное поражение, вовлечение проксимальных отделов конечностей, наличие чувствительных расстройств, диффузная демиелини-зация при ЭНМГ, белково-клеточная диссоциация в ликворе. Дифференциальная диагностика ММН и ХВДП крайне важна т. к. применяемый при лечении ХВДП преднизолон является не только неэффективным при ММН, но и согласно литературным данным может ухудшить состояние [5].
Лечение
С позиции доказательной медицины на сегодняшний день единственным средством лечения ММН является применение внутривенных иммуноглобулинов. Схема введения такова — 0,4 г/кг в течение 5 дней, либо 0,4 г/кг 1 раз в неделю 6 недель. В первые 2-4 недели становится заметно уменьшение слабости. Если начальное лечение иммуноглобулином является эффективным, то повторное лечение также следует проводить иммуноглобулином. Затем иммуноглобулин вводят по 0,4-2 г/кг каждый месяц, либо 0,2 г/кг 1 раз в неделю, а потом 1 раз в 2 недели. Доза и интервал введения определяются в зависимости от эффективности терапии. Однако, на фоне лечения значительному регрессу подвергаются лишь относительно недавно сформировавшиеся парезы, а длительно существовавшие дефекты остаются стабильными [4].
Затем иммуноглобулин вводят по 0,4-2 г/кг каждый месяц, либо 0,2 г/кг 1 раз в неделю, а потом 1 раз в 2 недели. Доза и интервал введения определяются в зависимости от эффективности терапии. Однако, на фоне лечения значительному регрессу подвергаются лишь относительно недавно сформировавшиеся парезы, а длительно существовавшие дефекты остаются стабильными [4].
Если терапия иммуноглобулином недостаточно эффективна или неэффективна, то может быть рассмотрена возможность иммуносупрессивной терапии, однако нет достоверных данных о ее эффективности. В качестве препарата используют циклофосфамид 1 г/кг в течение 6 месяцев в сочетании с двумя
процедурами плазмафереза. Однако токсичность делает цикло-фосфамид менее желательным вариантом. Применение корти-костероидной терапии не рекомендуется [13,14]. При частичной эффективности иммуноглобулинов, либо необходимости в их частом введении, возможно сочетание их циклофосфамидом, что позволяет удлинить интервал между инфузиями [4].
Клинический пример
Больной К, 34 лет, при поступлении в неврологическое отделение Ростовского гсударственного медицинского университета (04.2016 г.) предъявлял жалобы на ограничение объёма движений в левой руке (невозможность полного разгибания и сгибания пальцев руки в кулак), некоторое похудение левой кисти, слабость в ногах (больше в левой стопе), затруднение при ходьбе по ступенькам. Считает себя больным с апреля 2012 г., когда впервые появилась скованность, ограничение объема движений в пальцах левой кисти. Примерно через год присоединилась слабость в левой ноге, начал «пришлепывать». Еще через год отметил похудание мышц левой кисти. При появлении первых жалоб обратился к неврологу поликлиники, состояние расценено как проявление дегенеративного заболевания позвоночника, выполнена МРТ шейного отдела позвоночника, патологии не выявлено. Для исключения очагового поражения головного мозга, рекомендовано выполнение МРТ головного мозга — патологии не выявлено. На данном этапе симптоматика была расценена как проявления дизграфии, больному назначена сосудистая и нейротропная терапия. В связи с отсутствием улучшения и нарастанием симптоматики в течении последующего времени, вновь обратился к неврологу, госпитализирован в стационар по месту жительства, где курировался с хронической воспалительной демиелинизирующей полинейропатией, для уточнения диагноза направлен в РостГМУ.
На данном этапе симптоматика была расценена как проявления дизграфии, больному назначена сосудистая и нейротропная терапия. В связи с отсутствием улучшения и нарастанием симптоматики в течении последующего времени, вновь обратился к неврологу, госпитализирован в стационар по месту жительства, где курировался с хронической воспалительной демиелинизирующей полинейропатией, для уточнения диагноза направлен в РостГМУ.
Неврологический статус при поступлении: сознание ясное, пациент адекватен, ориентирован в пространстве и во времени.
Рисунок 1. Исследование левого малоберцового нерва при отведении с передней большеберцовой мышцы. Увеличение терминальной латентности 6,79 мс слева, 6 мс справа (при норме не более 4,5 мс). Определяется блок проведения.
Черепные нервы без патологии. Двигательная система: атрофия гипотенара левой кисти и межостных мышц кистей и стоп. Объем движений в проксимальных отделах конечностей полный, в дистальных отделах левой кисти и стопы ограничен. Мышечная сила в проксимальных отделах конечностей 5 баллов, дистально: в правой кисти — 5 баллов, в левой — 3,5 балла; мышечная сила в проксимальных отделах нижних конечностей — 5 баллов, дистально: в правой стопе — 5 баллов, в левой 3,5 балла. Мышечный тонус в верхних и нижних конечностях не изменен. Сухожильные рефлексы с верхних конечностей 8 = Б снижены, коленные 8 = Б снижены, ахилловы рефлексы отсутствует, патологических стопных знаков нет. Фасцикулярные подергивания мышц верхнего плечевого пояса при подъеме рук вверх. Убедительных данных за нарушение болевой, вибрационной чувствительности, мышечно-суставного чувства не выявлено. В позе Ромберга лёгкая шаткость без сторонности, ПНП, ПКП выполняет удовлетворительно, нистагма нет. Менингеальных знаков нет. Тазовые функции контролирует.
Объем движений в проксимальных отделах конечностей полный, в дистальных отделах левой кисти и стопы ограничен. Мышечная сила в проксимальных отделах конечностей 5 баллов, дистально: в правой кисти — 5 баллов, в левой — 3,5 балла; мышечная сила в проксимальных отделах нижних конечностей — 5 баллов, дистально: в правой стопе — 5 баллов, в левой 3,5 балла. Мышечный тонус в верхних и нижних конечностях не изменен. Сухожильные рефлексы с верхних конечностей 8 = Б снижены, коленные 8 = Б снижены, ахилловы рефлексы отсутствует, патологических стопных знаков нет. Фасцикулярные подергивания мышц верхнего плечевого пояса при подъеме рук вверх. Убедительных данных за нарушение болевой, вибрационной чувствительности, мышечно-суставного чувства не выявлено. В позе Ромберга лёгкая шаткость без сторонности, ПНП, ПКП выполняет удовлетворительно, нистагма нет. Менингеальных знаков нет. Тазовые функции контролирует.
Лабораторные показатели крови и мочи в пределах нормы, за исключением повышения уровня КФК — 296 МЕ/л. В ликворе обращало на себя внимание увеличение уровня белка до 0,82 г/л, положительные антитела к ганглиозиду GM-1.
В ликворе обращало на себя внимание увеличение уровня белка до 0,82 г/л, положительные антитела к ганглиозиду GM-1.
При выполнении стимуляционной ЭНМГ получены результаты свидетельствующие о наличии признаков дистальной демиелинизации подкрыльцовых, мышечно-кожных, лучевых, малоберцовых нервов с двух сторон в виде значительного повышения терминальной латентности. Обращает на себя внимание падение амплитуд и дисперсия моторных ответов при стимуляции проксимальных точек срединных нервов с двух сторон, что говорит и об их аксональном поражении. Также отмечается наличие выраженного блока проведения возбуждения (73,3%) по левому малоберцовому нерву (рис. 1).
На основании всего вышеизложенного был поставлен диагноз: Мультифокальная моторная нейропатия.
Данное наблюдение подтверждает, что диагностика мульти-фокальной моторной нейропатии является сложной, интервал «дебют — диагноз» составил 4 года. Значимую роль в дифференциальной диагностике имеет ЭНМГ, позволяющая установить аксонально-демиелинизирующее поражение нервов с преобладанием демиелинизиации, а также асимметричность вовлечения в патологический процесс нервных волокон.
Значимую роль в дифференциальной диагностике имеет ЭНМГ, позволяющая установить аксонально-демиелинизирующее поражение нервов с преобладанием демиелинизиации, а также асимметричность вовлечения в патологический процесс нервных волокон.
Мультифокальная моторная нейропатия относится к числу редких аутоиммунных демиелинизирующих полинейропатий, характеризующихся своеобразной клинической и электро-нейромиографической картиной. Учитывая схожесть клинической картины ММН с ХВДП и БАС, принимая во внимание высокую вероятность развития необратимой инвалидизации в случае отсутствия своевременной патогенетической терапии, необходимо знание диагностических критериев данной патологии и обязательное проведение ЭНМГ для своевременной постановки верного диагноза и назначения соответствующей терапии.
Литература
1. Ахмеджанова ЛТ. Клинико-иммунологическая характеристика хронических демиелинизирующих полиневропатий. : Автореферат диссертации канд. мед. наук: специальность 14.00.13 «нервные болезни». М., 2008: 29 с.
: Автореферат диссертации канд. мед. наук: специальность 14.00.13 «нервные болезни». М., 2008: 29 с.
2. Иосифова ОА. Моторная мультифокальная нейропатия (Клинико-нейрофизиологическое исследование): Автореферат диссертации канд. мед. наук: специальность 14.00.13 «нервные болезни», 14.00.16 «патологическая физиология». М., 2009: 29 с.
3. Кушнир ГМ, Иошина НН, Самохвалова ВВ. Международный неврологический журнал. 2014; 6(68): 93-98.
4. Левин ОС. Полинейропатии: Клиническое руководство. М.: ООО «Издательство «Медицинское информационное агентство»; 2016: 480 с.
5. Мультифокальная моторная нейропатия. В кн: Клинические рекомендации по неврологии. Европейской федерации неврологических сообществ. 2-е издание. М.: Издательский дом «АБВ-пресс»; 2012: 425-434.
6. Супонева НА. Клиническая и диагностическая роль аутоан-тител к ганглиозидам периферических нервов: обзор литературы и собственные данные. Нервно-мышечные болезни. 2013; 1: 26-34. DOI:10.17650/2222-8721-2013-0-1-26-34.
Супонева НА. Клиническая и диагностическая роль аутоан-тител к ганглиозидам периферических нервов: обзор литературы и собственные данные. Нервно-мышечные болезни. 2013; 1: 26-34. DOI:10.17650/2222-8721-2013-0-1-26-34.
7. Cornblath DR. Reserch criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1991; 41: 617-618. DOI:10.1212/wnl.41.5.617.
8. Donaghy AM. Multifocal motor neuropathy. Neurology India. 2002; 50: 408-416.
9. Gilhus NE, Barnes MP, Brainin M. Multifocal motor meuropathy. In European handbook of neurological management. 2nd еd. Wiley-Blackwell; Oxford; UK; 2010: Vol.1, ch. 21. 580 p. DOI:10.1002/9781444328394.
10 Jacobs BC, Rothbarth PH, van der Meche, Herbrink P, Schmitz PI, de Klerk MA, van Doorn PA. The spectrum of antecedent infections in Guillain-Barre syndrome: a case control study.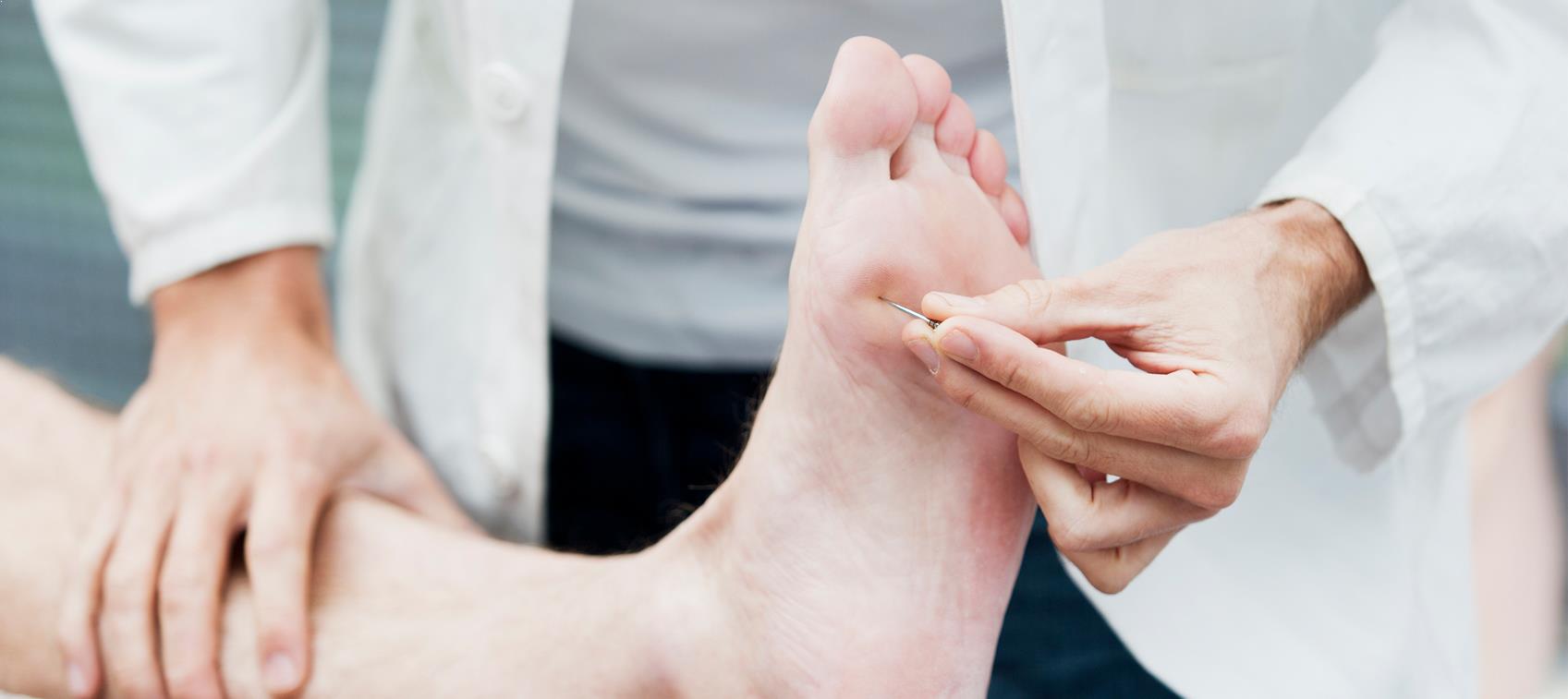 Neurology 1998; 51: 1110-1115. DOI:10.1212/WNL.51.4.1110
Neurology 1998; 51: 1110-1115. DOI:10.1212/WNL.51.4.1110
11. Kaji R, Kimura J. Facts and fallacies on anti-GM1 antibodies. Phisiology of motor neuropathies. Brain. 1999; 122: 797-798. DOI:10.1093/brain/122.5.797.
12. Lager JM, Gavriliuc E. Clinical differential diagnosis of multifocal motor neuropathy. European Neurological Review. 2012; 7(2): 124-127. DOI:10.17925/ENR.2012.07.02.124.
13. Meuth SG, Kleinschnitz С. Multifocal motor neuropathy: update on clinical characteristics, pathophysiological concepts and therapeutic options. Europian Neurology. 2010;63:193-204 DOI:10.1159/000282734
14 Nobile-Orazio E, Terenghi F, Carpo M, Cappellari A, Meucci N. Treatment of multifocal motor neuropathy. Neurological Science. 2003; 24: 251-255. DOI: 10.1007/s10072-003-0089-z
15.:max_bytes(150000):strip_icc()/GettyImages-508009390-5871d7d93df78c17b66b838f.jpg) O’Hanlon GM, Veitch J, Gallardo E, Paterson GJ. Peripheral neuropathy associated with anti-GM2 ganglioside antibodies: clinical and immunopathological studies. Autoimmunity. 2000;32:133-44. DOI:10.3109/08916930008994083.
O’Hanlon GM, Veitch J, Gallardo E, Paterson GJ. Peripheral neuropathy associated with anti-GM2 ganglioside antibodies: clinical and immunopathological studies. Autoimmunity. 2000;32:133-44. DOI:10.3109/08916930008994083.
16 Pestronk A, Cornblath DR, Ilyas AA, Adams RN, Clawson L. A treatable multifocal motor neropathy with antibodies to GM1 gan-gliosode. Ann Neurol 1988; 24: 73-80. DOI:10.1002/ana.410240113.
17. Shin Joh, Clinical electromyography: nerve conduction studies. 3th ed. Philadelphia: Lippincott Williams & Wilkins;2003. 851 p. DOI: 10.1016/S0387-7604(03)000808-3.
18 Van Schaik IN, Bossuyt PM, Brand A, Vermeulen M. Diagnostic value of GM1 antibodies in motor-neuron disorders and neuropathies: a meta-analysis. Neurology 1995;45:1570-7.
DOI:10.1212/WNL.45.8.1570.
References
1. Akhmedzhanova LT. Clinical and immunological characteristics of chronic demyelinating polyneuropathy: Abstract of the dissertation Cand. Med. Sciences: specialty 14.00.13 «Nervous diseases». Moscow, 2008: 30 р. (In Russ.)
2. Yosephova OA. Multifocal motor neuropathy (Clinical and neurophysiological study): abstract of thesis Cand. Med. Sciences: specialty 14.00.13 «Nervous diseases», 14.00.16 pathological physiology. Moscow, 2009: 29 p. (In Russ.)
3. Kushnir GM, Yoshina NN, Samokhvalova VV. International neurological journal. 2014; 6(68): 93-98 (In Russ.)
4. Levin OS. Polyneuropathy: Clinical manual. Moscow: OOO «Publisher «Medical information Agency»; 2016. 480 p. (In Russ.)
5. Multifocal motor neuropathy In. National clinical guidelines. 2th ed. Moscow: Publishing house «ABV-press»; 2012: 425-434. (In Russ.)
Multifocal motor neuropathy In. National clinical guidelines. 2th ed. Moscow: Publishing house «ABV-press»; 2012: 425-434. (In Russ.)
6. Suponeva ON. Clinical and diagnostic role of antibodies to gangliosides peripheral nerves: a literature review and own data. Neuro-muscular disease 2013; 1: 26-34.
DOI:10.17650/2222-8721-2013-0-1-26-34 (In Russ.)
7. Cornblath DR. Reserch criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1991; 41: 617-618. DOI:10.1212/wnl.41.5.617.
8. Donaghy AM. Multifocal motor neuropathy. Neurology India. 2002; 50: 408-416.
9. Gilhus NE, Barnes MP, Brainin M. Multifocal motor meuropathy. In European handbook of neurological management. 2nd еd. Wiley-Blackwell; Oxford; UK; 2010: Vol. 1, ch. 21. 580 p. DOI:10.1002/9781444328394.
1, ch. 21. 580 p. DOI:10.1002/9781444328394.
10 Jacobs BC, Rothbarth PH, van der Meche, Herbrink P, Schmitz PI, de Klerk MA, van Doorn PA. The spectrum of antecedent infections in Guillain-Barre syndrome: a case control study. Neurology 1998; 51: 1110-1115. DOI:10.1212/WNL.51.4.1110
11. Kaji R, Kimura J. Facts and fallacies on anti-GM1 antibodies. Phisiology of motor neuropathies. Brain. 1999; 122: 797-798. DOI:10.1093/brain/122.5.797.
12. Lager JM, Gavriliuc E. Clinical differential diagnosis of multifocal motor neuropathy. European Neurological Review. 2012; 7(2): 124-127. DOI:10.17925/ENR.2012.07.02.124.
13. Meuth SG, Kleinschnitz С. Multifocal motor neuropathy: update on clinical characteristics, pathophysiological concepts and therapeutic options. Europian Neurology. 2010;63:193-204 DOI:10. 1159/000282734
1159/000282734
14 Nobile-Orazio E, Terenghi F, Carpo M, Cappellari A, Meucci N. Treatment of multifocal motor neuropathy. Neurological Science. 2003; 24: 251-255. DOI: 10.1007/s10072-003-0089-z
15 O’Hanlon GM, Veitch J, Gallardo E, Paterson GJ. Peripheral neuropathy associated with anti-GM2 ganglioside antibodies: clinical and immunopathological studies. Autoimmunity. 2000;32:133-44. DOI:10.3109/08916930008994083.
16 Pestronk A, Cornblath DR, Ilyas AA, Adams RN, Clawson L. A treatable multifocal motor neropathy with antibodies to GM1 gan-gliosode. Ann Neurol 1988; 24: 73-80. DOI:10.1002/ana.410240113.
17. Shin Joh, Clinical electromyography: nerve conduction studies. 3th ed. Philadelphia: Lippincott Williams & Wilkins;2003. 851 p. DOI:10.1016/S0387-7604(03)000808-3.
18. Van Schaik IN, Bossuyt PM, Brand A, Vermeulen M. Diagnostic value of GM1 antibodies in motor-neuron disorders and neuropathies: a meta-analysis. Neurology 1995;45:1570-7. DOI:10.1212/WNL.45.8.1570.
Van Schaik IN, Bossuyt PM, Brand A, Vermeulen M. Diagnostic value of GM1 antibodies in motor-neuron disorders and neuropathies: a meta-analysis. Neurology 1995;45:1570-7. DOI:10.1212/WNL.45.8.1570.
Сведения об авторах
Гончарова Зоя Александровна — доктор медицинских наук, доцент, профессор кафедры нервных болезней и нейрохирургии, Ростовский государственный медицинский университет.
Адрес:Российская Федерация, 344022, г. Ростов-на-Дону, пер. Нахичеванский, 29; тел.: 8(909) 421.22.46; e-mail: [email protected].
Ковалева Наталия Сергеевна — кандидат медицинских наук, ассистент кафедры нервных болезней и нейрохирургии, Ростовский государственный медицинский университет.
Адрес:Российская Федерация, 344022, г. Ростов-на-Дону, пер. Нахичеванский, 29; тел.: 8(863) 2504200; e-mail: [email protected].
Нахичеванский, 29; тел.: 8(863) 2504200; e-mail: [email protected].
Authors
Goncharova Zoya Alexandrovna -MD, Dr. Med. Sci,. Prof., the Department of Neurology and Neurosurgery of the Rostov State Medical University.
Address: 29 Nakhichevansky str., Rostov-on-Don, 344022, Russian Federation 344022; phone 8(909) 421.22.46; e-mail: [email protected].
Kovaleva Natalia Sergeevna — MD, Cand Med Sci, Assistant of Professor of the Department of Neurology and Neurosurgery of the Rostov State Medical University.
Address: 29, Nakhichevansky str., Rostov-on-Don, 344022, Russian Federation 344022; phone 8(863)2504200; e-mail: [email protected].
Поступила 05.12.2016 Принята к печати 01.02.2017
Мультифокальная моторная нейропатия | Ведущие доктора | Лучшие клиники | Отзывы
Множественная или мультифокальная двигательная или моторная невропатия – это относительно редкое заболевание нервной системы, которое имеет предположительно аутоиммунный тип и характеризующееся демиелинизацией двигательных волокон. Заболевание опасно тем, что ведет к инвалидизации без активного адекватного лечения.
Заболевание опасно тем, что ведет к инвалидизации без активного адекватного лечения.
Лечение мультифокальной двигательной нейропатии в клиниках Германии, Израиля, Австрии, США, Финляндии и Швейцарии проводится при помощи комплекса медикаментозных средств включающих, в том числе, самые современные иммуномодуляторы.
Преимущества лечения мультифокальной моторной нейропатии за границей
Лечение мультифокальной моторной нейропатии за рубежом базируется на приеме иммуномодуляторов типа иммуноглобулина и циклофосфамида. Комплексное индивидуально сформированное лечение разработано таким образом, чтобы при максимальном воздействии минимизировать возможность проявления побочных эффектов. Кроме того активно применяются всевозможные продуктивные физиотерапевтические методики реабилитации.
Диагностика мультифокальной моторной нейропатии за границей
Пациенты рекомендуемых нами неврологических клиник для подтверждения и уточнения диагноза проходят все необходимые диагностические тесты, включая:
- электронейромиографию, обеспечивающую выявление множественных парциальных блоков проведения по двигательным волокна при наличии нормальных проведений по все еще чувствительным волокнам;
- биопсии выявляющей субклинические изменения в волокнах;
- лабораторных анализов крови;
- игольчатой электромиографии выявляющей признаки денервации.

Организация лечения мультифокальной моторной нейропатии за границей
Компания «Пациент Менеджмент» является лидером в оказании услуг по медицинскому менеджменту. Одной из наших сильных сторон является тесное сотрудничество с известными медицинскими центрами, занимающимися, в том числе, лечением неврологических заболеваний.
Мы организовываем лечение у опытных докторов, которые могут предложить своим иностранным пациентам высококвалифицированные услуги при адекватной стоимости лечения и великолепном сервисе. Очень важным плюсом сотрудничества с нашей компанией, является возможность сократить ожидание приема у необходимого специалиста до минимума.
Обращайтесь к нам в «Пациент Менеджмент», и организованное нами лечение мультифокальной двигательной нейропатии в лучших клиниках Германии, Израиля, Австрии, США, Финляндии или Швейцарии станет для Вас великолепным шансом на полное выздоровление!
Дистальная моторная нейропатия тип V (HMN5, дистальная спинальная амиотрофия)
Исследуемый материал Цельная кровь (с ЭДТА)
Метод определенияСеквенирование.
Выдаётся заключение врача-генетика!
Исследование мутаций в гене BSCL2.
Тип наследования.
Аутосомно-доминантный.
Гены, ответственные за развитие заболевания.
BSCL2 (BSCL2 GENE)
Ген расположен на хромосоме 11 в регионе 11q12.3. Содержит 11 экзонов.
Мутации в данном гене приводят также к развитию врожденной генерализованной липодистрофии тип 2, спастической параплегии тип 17 (синдром Сильвера).
Определение заболевания.
Дистальная моторная нейропатия V типа – наследственное медленно прогрессирующее мышечное заболевание, вызываемое дегенерацией нейронов передних рогов спинного мозга. На сегодняшний день известны два гена, ответственных за развитие HMN5A: BSCL2 и GARS.
Патогенез и клиническая картина.
Ген кодирует трансмембранный белок эндоплазматической сети сейпин. Мутации в нём приводят к нарушению гликозилирования сейпина, образованию и накоплению комплексов, приводящих к дегенерации мотонейронов. Первые признаки заболевания проявляются, как правило, в юношеском возрасте (16-17 лет), но начало заболевания может варьировать от 2 до 40 лет. Основными клиническими признаками являются слабость и гипотрофия мышц верхних конечностей, в особенности тенарной и межкостных мышц кисти. Заболевание прогрессирует медленно и в большинстве случаев ограничивается поражением дистальных отделов рук. У 40% больных отмечено распространение патологического процесса и на дистальные отделы ног. У 10% больных выявлено снижение вибрационной чувствительности в руках и наличие умеренно выраженной пирамидной симптоматики. При вовлечении нижних конечностей наблюдается слабость перонеальной мускулатуры, деформация стопы по типу pes cavus, потеря вибрационной чувствительности. Характерным признаком на электромиограме является отсутствие потенциала действия при наличии нормальных значений скоростей проведения импульса по периферическим нервам и дистальной латентности. Морфология характеризуется избыточной неравномерностью диаметра мышечных волокон.
Первые признаки заболевания проявляются, как правило, в юношеском возрасте (16-17 лет), но начало заболевания может варьировать от 2 до 40 лет. Основными клиническими признаками являются слабость и гипотрофия мышц верхних конечностей, в особенности тенарной и межкостных мышц кисти. Заболевание прогрессирует медленно и в большинстве случаев ограничивается поражением дистальных отделов рук. У 40% больных отмечено распространение патологического процесса и на дистальные отделы ног. У 10% больных выявлено снижение вибрационной чувствительности в руках и наличие умеренно выраженной пирамидной симптоматики. При вовлечении нижних конечностей наблюдается слабость перонеальной мускулатуры, деформация стопы по типу pes cavus, потеря вибрационной чувствительности. Характерным признаком на электромиограме является отсутствие потенциала действия при наличии нормальных значений скоростей проведения импульса по периферическим нервам и дистальной латентности. Морфология характеризуется избыточной неравномерностью диаметра мышечных волокон.
Частота встречаемости:
не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Horl, G., Malli, R., Reed, J. A., Dierick, I., Verpoorten, N., Warner, T. T., Proukakis, C., Van den Bergh, P., Verellen, C., Van Maldergem, L., Merlini, L., De Jonghe, P., Timmerman, V., Crosby, A. H., Wagner, K. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nature Genet. 36: 271-276, 2004.
- Ellsworth, R. E.; Ionasescu, V.; Searby, C.; Sheffield, V. C.; Braden, V. V.; Kucaba, T. A.; McPherson, J. D.; Marra, M. A.; Green, E. D.: The CMT2D locus: refined genetic position and construction of a bacterial clone-based physical map. Genome Res. 9: 568-574, 1999.
- Christodoulou, K.; Kyriakides, T.; Hristova, A. H.; Georgiou, D.-M.; Kalaydjieva, L.; Yshpekova, B.; Ivanova, T.; Weber, J. L.; Middleton, L. T.: Map- ping of a distal form of spinal muscular atrophy with upper limb predominance to chromosome 7p. Hum. Molec. Genet. 4: 1629-1632, 1995.
- OMIM
 H.; Georgiou, D.-M.; Kalaydjieva, L.; Yshpekova, B.; Ivanova, T.; Weber, J. L.; Middleton, L. T.: Map- ping of a distal form of spinal muscular atrophy with upper limb predominance to chromosome 7p. Hum. Molec. Genet. 4: 1629-1632, 1995.
H.; Georgiou, D.-M.; Kalaydjieva, L.; Yshpekova, B.; Ivanova, T.; Weber, J. L.; Middleton, L. T.: Map- ping of a distal form of spinal muscular atrophy with upper limb predominance to chromosome 7p. Hum. Molec. Genet. 4: 1629-1632, 1995. АУТОСОМНО-РЕЦЕССИВНАЯ ПЕРИФЕРИЧЕСКАЯ НЕЙРОПАТИЯ С НЕЙРОМИОТОНИЕЙ (ARAN-NM): ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ, ПОДТВЕРЖДЕННОГО МУТАЦИЕЙ В ГЕНЕ HINT1 | Клочкова
1. Говбах И.А. Современные подходы диагностики наследственных мото-сенсорных нейропатий // ScienceRise. — 2015. — Т. 3. — № 4 — С. 43–53. [Govbakh IA. Modern approaches to diagnostics of hereditary motor-sensory neuropathy. ScienceRise. 2015;3(4)43–53. (In Russ).] doi: 10.15587/2313-8416.2015.39134
2. Reilly MM, Murphy SM, Laura M. Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16(1):1–14. doi: 10.1111/j.1529-8027.2011.00324.x.
doi: 10.1111/j.1529-8027.2011.00324.x.
3. Saporta ASD, Sottile SL, Miller LJ, et al. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011;69(1):22–33. doi: 10.1002/ana.22166.
4. Lassuthova P, Brozkova DS, Krutova M, et al. Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics. 2015;16(1):43–54. doi: 10.1007/s10048-014-0427-8.
5. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009; 8(7):654–667. doi: 10.1016/S1474-4422(09)70110-3
6. Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80(12): 1304–1314. doi: 10.1136/jnnp.2008.158295.
J Neurol Neurosurg Psychiatry. 2009;80(12): 1304–1314. doi: 10.1136/jnnp.2008.158295.
7. Zhao H, Race V, Matthijs G, et al. Exome sequencing reveals HINT1 mutations as a cause of distal hereditary motor neuropathy. Eur J Hum Genet. 2014;22(6):847–850. doi: 10.1038/ejhg.2013.231.
8. Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6–14. doi: 10.1136/jnnp-2011-300952.
9. Zimon M, Baets J, Almeida-Souza L, et al. Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet. 2012;44(10):1080–1083. doi: 10.1038/ng.2406.
10. Aminkeng F. HINT1 mutations define a novel disease entity — autosomal recessive axonal neuropathy with neuromyotonia. Clin Genet. 2013;83(1):31–32. doi: 10.1111/cge.12030.
Clin Genet. 2013;83(1):31–32. doi: 10.1111/cge.12030.
11. Mertens HG, Zschocke S. [Neuromyotonia. (In German).] Klin Wochenschr. 1965;43(17):917–925. doi: 10.1007/BF01712058
12. Lance JW, Durke D, Pollard J. Neuromyotonia in the spinal form of Charcot-Marie-Tooth disease. Clin Exp Neurol. 1979;16:49–56.
13. Peeters K, Chamova T, Tournev I, Jordanova A. Axonal neuropathy with neuromyotonia: there is a HINT. Brain. 2017;140:868–877. doi: 10.1093/brain/aww301.
14. Hahn AF, Parkes AW, Bolton CF, Stewart SA. Neuromyotonia in hereditary motor neuropathy. J Neurol Neurosurg Psychiatry. 1991;54(3):230–235. doi: 10.1136/jnnp.54.3.230.
15. Black JT, Garcia-Mullin R, Good E, Brown S. Muscle rigidity in a newborn due to continuous peripheral nerve hyperactivity. Arch Neurol. 1972;27(5):413–425. doi: 10.1001/archneur.1972.00490170045007
Black JT, Garcia-Mullin R, Good E, Brown S. Muscle rigidity in a newborn due to continuous peripheral nerve hyperactivity. Arch Neurol. 1972;27(5):413–425. doi: 10.1001/archneur.1972.00490170045007
16. Auger RG, Daube JR, Gomez MR, Lambert EH. Hereditary form of sustained muscle activity of peripheral nerve origin causing generalized myokymia and muscle stiffness. Ann Neurol. 1984;15(1): 13–21. doi: 10.1002/ana.410150104.
17. Mcguire SA, Tomasovic JJ, Ackerman N. Hereditary Continuous Muscle-Fiber Activity. Arch Neurol. 1984;41(4):395–396. doi: 10.1001/archneur.1984.04050160057016
18. Grund G. [Zur Frage des Vorkommens erworbener Myotonie. (In German).] Dtsch Z Nervenheilkd. 1911;42(1–2):110–127. doi: 10.1007/bf01649723.
19. Grund G. [Uber genetische Beziehungen zwischen Myotonie, Muskelkrampfen und Myokymie. (In German).] Dtsch Z Nervenheilkd. 1938;146(1–2):3–14. doi: 10.1007/bf01762426.
Grund G. [Uber genetische Beziehungen zwischen Myotonie, Muskelkrampfen und Myokymie. (In German).] Dtsch Z Nervenheilkd. 1938;146(1–2):3–14. doi: 10.1007/bf01762426.
20. Gamstorp I, Wohlfart G. A syndrome characterized by myokymia, myotonia, muscular wasting and increased perspiration. Acta Psychiatr Scand. 1959;34(2):181–194. doi: 10.1111/j.1600-0447.1959.tb07573.x.
21. Vasilescu C, Alexianu M, Dan A. Neuronal type of Charcot-Marie-Tooth Disease with a syndrome of contrinuous motor unit activity. J Neurol Sci. 1984;63(1):11–25. doi: 10.1016/0022-510x(84)90105-9.
22. Zimon M, Battaloglu E, Parman Y, et al. Unraveling the genetic landscape of autosomal recessive Charcot-Marie-Tooth neuropathies using a homozygosity mapping approach. Neurogenetics. 2014;16(1):33–42. doi: 10.1007/s10048-014-0422-0.
doi: 10.1007/s10048-014-0422-0.
23. Мальмберг С.А., Куренков А.Л. Аксональная моторная полиневропатия с гиперактивностью двигательных единиц // Неврологический журнал. — 2002. — Т. 7. — № 6 — С. 28–33. [Mal’mberg SA, Kurenkov AL. Aksonal’naya motornaya polinevropatiya s giperaktivnost’yu dvigatel’nykh edinits. Journal of neurology. 2002;7(6):28–33. (In Russ).]
24. Никитин С.С., Куренков А.Л. Методические основы транскраниальной магнитной стимуляции в неврологии и психиатрии. — М.: «ИПЦ Маска»; 2006. — С. 160–166. [Nikitin SS, Kurenkov AL. Metodicheskie osnovy transkranial’noi magnitnoi stimulyatsii v nevrologii i psikhiatrii. Moscow: Maska; 2006. p. 160–166. (In Russ).]
25. Barbier E, Zapata A, Oh E, et al. Supersensitivity to amphetamine in protein kinase-C interacting protein/HINT1 knockout mice. Neuropsychopharmacology. 2007;32(8):1774–1782. doi: 10.1038/sj.npp.1301301.
Neuropsychopharmacology. 2007;32(8):1774–1782. doi: 10.1038/sj.npp.1301301.
26. Liu Q, Puche AC, Wang JB. Distribution and expression of protein kinase c interactive protein (PKCI/HINT1) in mouse central nervous system (CNS). Neurochem Res. 2008;33(7):1263–1276. doi: 10.1007/s11064-007-9578-4.
27. Caetano JS, Costa C, Baets J, et al. Autosomal recessive axonal neuropathy with neuromyotonia: a rare entity. Pediatr Neurol. 2014;50(1):104–107. doi: 10.1016/j.pediatrneurol. 2013.08.028.
28. Jerath NU, Shy ME, Grider T, Gutmann L. A case of neuromyotonia and axonal motor neuropathy: a report of a HINT1 mutation in the United States. Muscle Nerve. 2015;52(6):1110-1113. doi: 10.1002/mus.24774.
29. Rauchenzauner M, Fruhwirth M, Hecht M, et al. A novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: thorough neurological examination gives the clue. Neuropediatrics. 2016;47(2):119–122. doi: 10.1055/s-0035-1570493.
Rauchenzauner M, Fruhwirth M, Hecht M, et al. A novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: thorough neurological examination gives the clue. Neuropediatrics. 2016;47(2):119–122. doi: 10.1055/s-0035-1570493.
30. Boaretto F, Cacciavillani M, Mostacciuolo ML, et al. Novel loss-of-function mutation of the HINT1 gene in a patient with distal motor axonal neuropathy without neuromyotonia. Muscle Nerve. 2015;52(4):688–689. doi: 10.1002/mus.24720.
31. Вавилов М.А., Бландинский В.Ф., Громов И.В. и др. Артродезирующие операции у детей старше 10 лет с деформациями стоп различной этиологии // Гений ортопедии. — 2016. — № 3 — С. 35–38. [Vavilov MA, Blandinskii VF, Gromov IV, et al. Arthrodesing surgeries in children above 10 years of age with feet deformities of various etiologies./cdn.vox-cdn.com/uploads/chorus_image/image/47221806/o-REFLEX-TEST-facebook.0.0.jpg) Genij ortopedii. 2016;(3):35–38. (In Russ).] doi: 10.18019/1028-4427-2016-3-35-38.
Genij ortopedii. 2016;(3):35–38. (In Russ).] doi: 10.18019/1028-4427-2016-3-35-38.
Мультифокальная моторная нейропатия — Неврология — LiveJournal
Актуальность. Из-за трудностей диагностики и схожести клинических проявлений мультифокальной моторной нейропатии (ММН) с хронической воспалительной демиелинизирующей полинейропатией (ХВДП) и боковым амиотрофическим склерозом (БАС), данная нозология требует повышенного внимания, поскольку, в отличии от БАС, не является фатальным заболеванием и при своевременно начатой терапии не приводит к необратимой инвалидизации. В то же время тактика лечения таких больных кардинально отличается от таковой при ХВДП, что в свою очередь диктует необходимость повышенной настороженности при обследовании больных с подозрением на демиелинизирующие полинейропатии.читайте также пост: Хроническая воспалительная демиелинизирующая полинейропатия (на laesus-de-liro.
 livejour nal.com) [читать]
livejour nal.com) [читать]читайте также пост: Боковой амиотрофический склероз (на laesus-de-liro.livejournal.com) [читать]
ММН — отдельная форма дизиммунных заболеваний периферической нервной системы, которая характеризуется локальной демиелинизацией [преимущественно] [!!!] моторных волокон (с вторичной аксонопатией), проявляющейся асимметричной прогрессирующей слабостью (и нейротрофическими расстройствами) преимущественно в дистальных группах мышц верхних конечностей (без чувствительных нарушений или с минимально выраженными чувствительными расстройствами).
Частота встречаемости ММН в популяции невелика и составляет 1 — 2 случая на 100 000 населения, чаще страдают мужчины, в среднем соотношение мужчины : женщины составляет 3 : 1. Дебют заболевания приходится на трудоспособный возраст (в среднем 40 лет), до 80% больных находятся в возрасте 20 — 50 лет).
В основе ММН лежит избирательная демиелинизация моторных волокон (вероятно, вследствие разного антигенного состава двигательных и чувствительных волокон), возникающая в ответ на аутоиммунную атаку против миелина. ММН ассоциирована с увеличением уровня антител класса М к ганглиозиду GM-1 в крови и отличается наличием ответа на терапию иммуно-модуляторами. По всей вероятности, вследствие этого при ММН имеется стойкая демиелинизация периферических нервов, так называемые блоки проведения возбуждения. Стойко сохраняющаяся локальная демиелинизация вызывает вторичные аксональные нарушения и нейротрофические расстройства. На сегодняшний день остается неясной причина селективного поражения двигательных волокон, не исключено, что так происходит , либо их различной чувствительности к повреждающим факторам.
читайте также пост: Ганглиозиды — биохимия и нейроиммунология (на laesus-de-liro.livejournal.com) [читать]
Заболевание начинается у большинства пациентов остро, однако, в ряде случаев возможно подострое развитие. Течение носит медленно либо скачкообразно прогрессирующий характер. Основное проявление ММН — формирование асимметричных парезов мышц конечностей, преимущественно в дистальных отделах, из-за поражения нескольких периферических нервов. Слабость в дистальных отделах рук встречается гораздо чаще, чем ног, и выявляется примерно у 95% пациентов с MMН. Чаще всего страдают локтевой, срединный, лучевой, малоберцовый и большеберцовый нервы. Слабость разгибания отдельных пальцев является распространенным ранним симптомом. Более чем у половины больных отмечаются фасцикуляции и крампи, из-за чего могут возникнуть ложные предположения о наличии у пациента бокового амиотрофического склероза (БАС). Сухожильные рефлексы меняются асимметрично, снижаясь либо выпадая с паретичных мышц и оставаясь не измененными с интактных.
читайте также пост: Крампи — болезненные мышечные спазмы (на laesus-de-liro.livejournal.com) [читать]
Чувствительные расстройства при ММН отсутствуют, однако у некоторых больных могут быть жалобы на онемение либо парестезии. Атрофии мышц формируются достаточно медленно, по мере развития вторичной аксональной дегенерации. Однако, иногда атрофия может развиваться достаточно быстро (еще до развития значимого аксонального повреждения), что связывают с нарушением нейротрофического обмена между нервом и мышцей из-за накопления в синаптической щели аутоантител. Поражение вегетативной нервной системы, черепных нервов, нервов иннервирующих дыхательную мускулатуру не характерно. Степень инвалидизации в целом коррелирует с продолжительностью заболевания.
Рабочая группа экспертов разработала клинические критерии диагностики ММН. К ключевым критериям относятся медленно или скачкообразно прогрессирующая локальная асимметричная слабость в конечностях, сохраняющаяся более месяца. Двигательные расстройства выявляются не менее чем в двух двигательных нервах. Если симптомы распространяются только на 1 двигательный нерв, то ставится вероятный диагноз. Незначительные чувствительные расстройства в дебюте заболевания (но могут развиваться с течением времени). Критериями исключения являются признаки поражения верхнего мотонейрона, бульбарные расстройства, чувствительные нарушения более явные, чем незначительное снижение вибрационной чувствительности на ногах и диффузная симметричная слабость в течение первых недель от начала заболевания (подробнее в статье №2 — см. раздел «Литература»).
Золотым стандартом в диагностике ММН является электронейромиография (ЭНМГ), в частности стимуляционная, которая обнаруживает множественные парциальные блоки проведения по двигательным волокнам (моторные блоки проведения — МБП) вне мест типичной компрессии, при нормальном проведении по сенсорным волокнам. Для более точного определения пораженного участка, шаг исследования нерва вместо обычных 10 см составляет 1-2 см. Из-за наличия блока проведения развивается снижение амплитуды либо площади М-ответа, полученного при стимуляции проксимального отдела нерва, относительного такового при более дистальной его стимуляции.
При исследовании лабораторных показателей у больных с ММН следует обратить внимание, что возможно незначительное увеличение [1] уровня белка в ликворе, повышение [2] уровня креатинфосфокиназы (КФК) в крови и повышение [3] титра IgM-аутоантител к GM1-ганглиозиду.
читайте также пост: Креатинкиназа (справочник невролога) (на laesus-de-liro.livejournal.com) [читать]
Обратите внимание! Учитывая, что, согласно данным литературы, частота выявляемости анти-GM1 IgM при ММН варьирует от 30 до 80%, отрицательные результаты иммунологического исследования при наличии других критериев данный диагноз не исключают. Таким образом, в настоящее время обнаружение повышения уровня анти-GM1-аутоантител является вспомогательным диагностическим критерием при данном заболевании, оказывающим помощь в постановке окончательного диагноза в сложных случаях. Эти антитела, однако, не являются диагностическим маркером для ММН, так как они могут определяться у больных с другими иммуноопосредованными нейропатиями.
В последние годы при исследовании пациентов с ММН большое внимание уделяют таким методам визуализации, как магнитно-резонансная томография (МРТ) и ультразвуковое исследование (УЗИ) периферических нервов. При ММН оба метода выявляют фокальное асимметричное утолщение [1] периферических нервов и [2] нервов плечевого сплетения с преимущественным увеличением нервных структур в проксимальных отделах.
Обратите внимание! Диагноз ММН ставится на основании клинических критериев (см. выше) Европейской федерации неврологических обществ (European Federation of Neurological Societies, EFNS) и обнаружения блоков проведения в двигательных нервах.
С позиции доказательной медицины на сегодняшний день единственным средством лечения ММН является применение внутривенных иммуноглобулинов. Схема введения: 0,4 г/кг в течение 5 дней, либо 0,4 г/кг 1 раз в неделю 6 недель (в первые 2 — 4 недели становится заметно уменьшение слабости; если начальное лечение иммуноглобулином является эффективным, то повторное лечение также следует проводить иммуноглобулином), затем иммуноглобулин вводят по 0,4 — 2 г/кг каждый месяц, либо 0,2 г/кг 1 раз в неделю, а потом 1 раз в 2 недели. Доза и интервал введения определяются в зависимости от эффективности терапии. Однако, на фоне лечения значительному регрессу подвергаются лишь относительно недавно сформировавшиеся парезы, а длительно существовавшие дефекты остаются стабильными. При частичной эффективности иммуноглобулинов, либо необходимости в их частом введении, возможно сочетание их циклофосфамидом (см. далее), что позволяет удлинить интервал между инфузиями. Если терапия иммуноглобулином недостаточно эффективна или неэффективна, то может быть рассмотрена возможность иммуносупрессивной терапии (однако нет достоверных данных о ее эффективности). При этом используют циклофосфамид 1 г/кг в течение 6 месяцев в сочетании с двумя процедурами плазмафереза. Однако токсичность делает циклофосфамид менее желательным вариантом. Применение кортикостероидной терапии не рекомендуется.
Литература:
статья «Мультифокальная моторная нейропатия» Гончарова З. А., Ковалева Н. С.; Ростовский государственный медицинский университет, Ростов-на-Дону (журнал «Сибирское медицинское обозрение» №1, 2017) [читать];
статья «Моторная мультифокальная нейропатия с блоками проведения (обзор литературы и описание двух клинических случаев)» Кушнир Г.М., Иошина Н.Н., Самохвалова В.В., Кузина О.С., Крылова А.Н., Сидоренко Н.А., Ибрагимова Л.Р.; ГУ «Крымский государственный медицинский университет им. С.И. Георгиевского»; 7-я Городская клиническая больница; КРУ КТМО «Университетская клиника» г. Симферополь, АР Крым (Международный неврологический журнал, №6, 2014) [читать];
статья «Мультифокальная моторная невропатия» Ю. В. Тринитатский, К. А. Острова, Т. В. Сычева, Т. И. Кушнаренко; ГБУ РО «Ростовская областная клиническая больница», г. Ростов-на-Дону; (журнал «Главный врач» №4, 2017) [читать];
статья «Сложности диагностики мультфокально моторной нейропатии» О.И. Чижевская, Е.А. Марусиченко; Институт неотлож-ной и восстановительно хирургии им. В.К. Гусака, г. Донецк (журнал «Архив клинической и экспериментальной медицины» №1, 2017) [читать];
статья «Мультифокальная моторная нейропатия с блоками проведения» А.В. Красильников, В.А. Наймушин; ГБУ «Республи-канская клиническая больница», Йошкар-Ола, Россия (Журнал неврологии и психиатрии, №6, 2016) [читать];
статья «Ультразвуковая визуализация периферических нервов при мультифокальной моторной нейропатии и хронической воспалительной демиелинизирующей полинейропатии» Д.С. Дружинин, Е.С. Наумова, С.С. Никитин; ГБОУ ВПО «Ярославский государственный медицинский университет» МЗ РФ, Ярославль; Региональная общественная организация «Общество специа-листов по нервно-мышечным болезням», Медицинский центр «Практическая неврология», Москва (журнал «Нервно-мышечные болезни» №1, 2016) [читать];
статья «Оценка динамики площади поперечного сечения периферических нервов при мультифокальной моторной нейропатии по данным ультразвукового исследования на фоне внутривенной терапии иммуноглобулинами: описание клинического случая» С.С. Никитин, Е.С. Наумова, Д.С. Дружинин; Региональная общественная организация «Общество специалистов по нервно-мышечным болезням», Медицинский центр «Практическая неврология», Москва; ФГБОУ ВО «Ярославский государственный медицинский университет» МЗ РФ, Ярославль (журнал «Нервно-мышечные болезни» №4, 2016) [читать];
статья «Мультифокальная моторная невропатия без блоков проведения и антител к ганглиозидам класса GM1» А.Г. Санадзе, Л.Ф. Касаткина, О.В. Гильванова, С.А. Иванов; Московский миастенический центр (Журнал неврологии и психиатрии, №8, 2011) [читать];
описание изобретения к патенту «Способ диагностики мультифокальной моторной невропатии» Суслина З.А., Вуйцик Н.Б, Ахмеджанова Л.Т., Строков И.А. ФГБУ «НЦН» РАМН, 2013 [читать];
автореферат диссертации на соискание ученой степени к.м.н. «Моторная мультифокальная нейропатия (клинико-нейро-физиологическое исследование)» Иосифова О.А., Московский государственный медико-стоматологический университет, Москва, 2009 [читать];
статья «Внутривенная высокодозная иммунотерапия: практические рекомендации по применению в лечении дизиммунных заболеваний периферического нейромоторного аппарата» Н.А. Супонева, Д.А. Гришина; ФГБНУ «Научный центр неврологии», Москва (журнал «Нервно-мышечные болезни» №4, 2015) [читать];
статья «Современные представления об идиопатических воспалительных полинейропатиях» О.Л. Пелехова, Харьковская медицинская академия последипломного образования (Международный медицинский журнал, №4, 2012) [читать];
статья «Ультразвуковая семиотика периферических нервов при мультифокальной моторной невропатии и боковым амио-трофическим склерозом (краткое сообщение)» Вуйцик Н.Б., Завалишин И.А., Ахмеджанова Л.Т., Строков И.А.; ФГБУ «Научный центр неврологии» РАН; ГБОУ ВПО «Первый Московский государственный медицинский университет им. И. М. Сеченова» МЗ РФ, Москва (журнал «Радиология-Практика» №2, 2014) [читать];
статья «Ультразвуковые изменения периферических нервов при мультифокальной моторной невропатии» Н.Б. Вуйцик, И.А. Строков, Г.И. Кунцевич, Л.Т. Ахмеджанова, З.А. Cуслина, Н.Н. Яхно; ФГБУ «Научный центр неврологии» РАН, Москва; ГБОУ ВПО «Первый Московский государственный медицинский университет им. И. М. Сеченова» МЗ РФ, Москва (Неврологический журнал, №1, 2014) [читать];
статья «Синдром верхнего вялого парапареза при БАС и БАС-подобных синдромах: вопросы дифференциальной диагностики» М.Н. Захарова, И.В. Закройщикова, И.С. Бакулин, И.А. Кочергин; ФГБНУ Научный центр неврологии, Москва (журнал «Medica Mente» №1, 2016) [читать]
Мультифокальная моторная нейропатия: причины, симптомы и лечение
Что такое мультифокальная моторная нейропатия?
Мультифокальная моторная нейропатия (MMN) — это заболевание, поражающее двигательные нервы вашего тела. Это нервы, которые контролируют ваши мышцы. Из-за этого состояния им трудно посылать электрические сигналы, которые двигают ваше тело, из-за чего ваши руки и руки кажутся слабыми. Они также будут дергаться и сводить судороги.
MMN не опасен для жизни, и в большинстве случаев лечение может укрепить мышцы.Заболевание может постепенно ухудшаться, и через некоторое время вам, возможно, станет трудно выполнять повседневные задачи, например печатать или одеваться. Но у многих людей симптомы могут быть настолько легкими, что они вообще не нуждаются в лечении. После постановки диагноза вы сможете работать и оставаться активными в течение многих лет.
Большинство людей получают диагноз MMN в возрасте от 40 до 50 лет, хотя взрослые в возрасте от 20 до 80 лет могут узнать, что у них это заболевание.
Причины
Никто не знает, что вызывает MMN. Ученые знают, что это аутоиммунное заболевание, то есть ваша иммунная система по ошибке атакует нервные клетки, как если бы они были захватчиками.Исследователи изучают болезнь, чтобы попытаться выяснить, почему она возникает.
Симптомы
Если у вас MMN, вы, скорее всего, заметите первые симптомы в руках и предплечьях. Ваши мышцы могут ощущаться слабыми, сокращаться или подергиваться так, что вы не можете это контролировать. Это может начаться в определенных частях руки или кисти, например, на запястье или пальце. Обычно симптомы более серьезны на одной стороне тела. Со временем болезнь может поразить и ваши ноги.
MMN безболезненно, и вы все равно сможете чувствовать руками, потому что ваши сенсорные нервы не затронуты.Но с возрастом ваши симптомы будут постепенно ухудшаться.
Получение диагноза
Врачи часто принимают MMN за боковой амиотрофический склероз (БАС), также известный как болезнь Лу Герига. У них похожие симптомы, например, подергивание. Однако, в отличие от БАС, MMN поддается лечению.
Ваш врач может посоветовать вам обратиться к неврологу, специалисту, который лечит проблемы с нервной системой. Вам проведут медицинский осмотр, а также зададут вопросы о ваших симптомах, например:
- Какие мышцы вызывают у вас проблемы?
- На одной стороне тела хуже?
- Как давно вы так себя чувствуете?
- Есть ли у вас онемение или покалывание?
- Облегчает ли что-нибудь симптомы? Что делает их хуже?
Врач проведет несколько анализов нервов и крови, чтобы исключить другие состояния, которые могут вызывать ваши симптомы.Они могут сделать:
- Исследование нервной проводимости (NCS). Этот тест измеряет, насколько быстро электрические сигналы проходят через ваши нервы. Обычно ваш врач помещает два датчика на кожу над одним из ваших нервов: один для передачи небольшого электрического разряда, а другой для регистрации активности. Врач повторит тест на других нервах, если посчитает, что задействовано несколько нервов.
- Игольчатая электромиография (ЭМГ). Ваш врач наложит электроды вам на руки. У электродов есть маленькие иглы, которые входят в ваши мышцы, и они прикреплены проводами к устройству, которое может измерять электрическую активность в ваших мышцах.Врач попросит вас медленно согнуть и расслабить руки, чтобы машина могла записывать активность. Врач может провести этот тест одновременно с NCS.
- Анализ крови на антитела к GM1, часть иммунной системы вашего организма. У некоторых людей с MMN их уровень выше. Если у вас действительно много этих антител, скорее всего, у вас есть болезнь. Однако у вас может быть MNN, даже если у вас мало антител.
Вопросы к врачу
- Как MMN повлияет на меня?
- Нужно ли мне лечение?
- Какие бывают виды?
- Что они заставят меня чувствовать?
- Смогу ли я работать?
- Что будет, если лечение не поможет?
- Могу ли я принять участие в каких-либо клинических испытаниях?
- Есть какие-то действия, которые я не смогу делать?
Лечение
Если у вас очень легкие симптомы, возможно, вам не потребуется никакого лечения.Если вам действительно нужно лечение, ваш врач, вероятно, назначит лекарство, называемое внутривенным иммуноглобулином (IVIg). Вы введете лекарство прямо в одну из своих вен через капельницу. Обычно вы получаете его в кабинете врача, хотя вы можете научиться делать это дома.
Если внутривенный иммуноглобулин работает, вы почувствуете, что ваша мышечная сила улучшится в течение 3–6 недель. Однако со временем эффекты исчезнут, поэтому вам нужно будет продолжать лечение. Обычно люди проходят это один раз в месяц, но это может быть более или менее частым в зависимости от вашего состояния.
IVIg не имеет большого количества побочных эффектов, но стоит дорого.
Врачи проверяют, как вводить препарат непосредственно через кожу, как обычную инъекцию, но этот метод доступен не всем.
Если внутривенный иммуноглобулин не работает, ваш врач может попробовать лекарство под названием циклофосфамид (цитоксан), которое также используется для лечения определенных типов рака. Этот препарат контролирует симптомы, подавляя вашу иммунную систему. В отличие от иммуноглобулина, циклофосфамид может иметь серьезные побочные эффекты, поэтому врачи стараются его не использовать.
Забота о себе
Если вы начнете лечение от MMN на ранней стадии, у вас, скорее всего, будет немного симптомов или возникнут долгосрочные проблемы. Придерживайтесь своего плана лечения и поговорите со своим врачом о любых изменениях в своем самочувствии.
Если у вас есть проблемы с определенными видами деятельности, вы можете обратиться к эрготерапевту или физиотерапевту. Они могут помочь вам сохранить мышцы сильными и показать, как легче выполнять повседневные задачи, если мышцы вас беспокоят.
Чего ожидать
Многие люди с MMN могут продолжать большую часть или хотя бы часть своих обычных действий.У некоторых людей болезнь может ухудшиться, и они не смогут выполнять повседневные дела.
Тип ваших проблем зависит от того, какие мышцы поражены. Если у вас слабые мышцы рук, у вас могут возникнуть проблемы с едой, набором текста, письмом или застегиванием одежды. Если затронуты мышцы ног, у вас могут возникнуть проблемы с ходьбой. У некоторых людей с тяжелой формой MMN есть проблемы во всех этих областях.
Мультифокальная моторная нейропатия — NORD (Национальная организация редких заболеваний)
УЧЕБНИК
Шайк И.Н., Леже Л.М., Нобиле-Орацио Э. и др.Мультифокальная моторная нейропатия. В: European Handbook of Neurological Management: Volume 1, 2nd ed., Gilhus NE, Barnes MP, Brainin M, editors. 2011 г .; Blackwell Publishing Ltd, Оксфорд, Великобритания. Стр. 343-350.
https://www.eaneurology.org/fileadmin/user_upload/guidline_papers/EFNS_guideline_2011_Multifocal_motor_neuropathy.pdf
СТАТЬИ ЖУРНАЛА
Kumar A, Patwa HS, Nowak RJ. Иммуноглобулинотерапия в лечении мультифокальной моторной нейропатии. J Neurol Sci. 2017; 375: 190-197. https: // www.ncbi.nlm.nih.gov/pubmed/28320129
Филиберт М., Грапперон А.М., Дельмонт Э., Аттариан С. Мониторинг краткосрочного эффекта внутривенных иммуноглобулинов при мультифокальной моторной нейропатии с использованием индекса числа двигательных единиц. Clin Neurophysiol. 2017; 128: 235-240. https://www.ncbi.nlm.nih.gov/pubmed/27988478
Тобон А. Роль иммуноглобулина в лечении иммуноопосредованных периферических невропатий. J Infus Nurs. 2017; 40: 375-379. https://www.ncbi.nlm.nih.gov/pubmed/2
86
Stangel R, Gold R, Pittrow D, et al.Лечение пациентов с мультифокальной моторной нейропатией иммуноглобулинами в клинической практике: регистр SIGNS. Ther Adv Neurol Disord. 2016; 9: 165-179. https://www.ncbi.nlm.nih.gov/pubmed/27134672
Ishigaki H, Hiraide T, Miyagi Y, et al. Мультифокальная моторная нейропатия с началом в детстве с антителами иммуноглобулина М к ганглиозидам GM1 и GM2: отчет о клиническом случае и обзор литературы. Pediatr Neurol. 2016; 62: 51-57. https://www.ncbi.nlm.nih.gov/pubmed/27400822
Lawson VH, Arnold WD.Мультифокальная моторная нейропатия: обзор патогенеза, диагностики и лечения. Neuropsychiatr Dis Treat. 2014; 10: 567-576. https://www.ncbi.nlm.nih.gov/pubmed/24741315
Van Asseldonk JT, Franssen H, Van den Berg-Vos RM, Wokke JH, Van den Berg LH. Мультифокальная моторная нейропатия. Lancet Neurol. 2005; 4: 309-319. https://www.ncbi.nlm.nih.gov/pubmed/15847844
Van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Мультифокальная моторная нейропатия: долгосрочная клиническая и электрофизиологическая оценка поддерживающего лечения внутривенным иммуноглобулином.Головной мозг. 2002; 125 (часть 8): 1875-1886. https://www.ncbi.nlm.nih.gov/pubmed/12135977
Van den Berg LH, Franssen H, Wolke JH. Долгосрочный эффект лечения внутривенным иммуноглобулином при мультифокальной моторной нейропатии. Головной мозг. 1998; 121 (Pt3): 421-428. https://www.ncbi.nlm.nih.gov/pubmed/9549518
Lange DJ, Trojaborg W, Latov N, et al. Мультифокальная моторная нейропатия с блокадой проводимости: это отдельная клиническая форма? Неврология 1992; 42: 497-505.
ИНТЕРНЕТ
Ланге Д., Робинсон-Папп Дж.Мультифокальная моторная нейропатия. UpToDate, Inc., 2017 г. 17 июля. Доступно по адресу: https://www.uptodate.com/contents/multifocal-motor-neuropathy, дата обращения 25 апреля 2018 г.
Ланге Д., Робинсон-Папп Дж. Иммуноопосредованные невропатии. UpToDate, Inc., 2017 г. 17 июля. Доступно по адресу: https://www.uptodate.com/contents/immune-mediated-neuropathies, дата обращения 25 апреля 2018 г.
Национальный институт неврологических расстройств и инсульта. Мультифокальная моторная нейропатия. 3 марта 2016 г. Доступно по адресу: https: // rarediseases.info.nih.gov/diseases/11011/multifocal-motor-neuropathy По состоянию на 26 апреля 2018 г.
Мультифокальная моторная нейропатия (MMN) — Международный фонд GBS / CIDP
Что такое MMN?
MMN (мультифокальная моторная нейропатия) — редкое заболевание, при котором фокальные области множественных моторных нервов атакуются собственной иммунной системой. Обычно MMN медленно прогрессирует, что приводит к асимметричной слабости конечностей пациента. У пациентов часто появляется слабость в руке (-ях), что приводит к падению предметов или иногда неспособности повернуть ключ в замке.Слабость, связанная с MMN, может быть определена как соответствие определенной нервной территории. По сути, нет онемения, покалывания или боли. Пациенты с MMN могут иметь другие симптомы, включая подергивание или небольшие случайные ямочки на мышцах под кожей, которые неврологи называют фасцикуляциями.
Клиническое течение MMN хронически прогрессирующее, без ремиссии. Распространенность этого очень редкого заболевания оценивается в 0,6 случая на 100 000 человек, что делает его еще более редким, чем СГБ, спонтанно самоизлечивающееся заболевание, при котором 1-2 случая на 100 000 ежегодно возникают в Северной Америке и Европе.
MMN имеет много функций, аналогичных CIDP, в том, что его начало прогрессирует с течением времени, вызывая повышенную инвалидность, которая отражает большее количество вовлеченных нервных участков. Однако, в отличие от CIDP, MMN асимметричен и по-разному влияет на правую и левую стороны тела. Считается, что в отличие от других форм хронического воспаления нервов он никогда не проходит. Однако соответствующее лечение, одобренное FDA, ограничивает прогрессирование и улучшает функцию.
Что вызывает MMN?
MMN связан с повышенным уровнем специфических антител к GM1, ганглиозиду или сахаросодержащему липиду, обнаруженному в периферических нервах.Антитела обычно защищают людей от вирусов и бактерий, но при определенных обстоятельствах могут связываться и способствовать иммунной атаке на периферический нерв. Эти антитела были обнаружены с помощью новейших анализов почти у всех пациентов с MMN. Даже если эти антитела не вызывают повреждения нервов, они могут быть важным маркером заболевания и облегчать диагностику.
Как диагностируется MMN?
Диагноз MMN является клиническим, который зависит от подтверждения того, что пациент:
- имеет чисто двигательное расстройство, затрагивающее отдельные нервы
- нет признаков UMN (верхних двигательных нейронов), таких как оживленные рефлексы в коленях или лодыжках или спастичность в конечностях
- нет проблем с речью или глотанием
- нет сенсорный дефицит
- есть свидетельства наличия фокальных областей нерва, в которых электрические импульсы замедляются или блокируются (блокировка проводимости), что может быть обнаружено с помощью электрофизиологических тестов
Эти критерии разработаны для дифференциации MMN от ALS (болезнь Лу Герига), Синдром Льюиса-Самнера (мультифокальное воспалительное заболевание нервов с двигательными и сенсорными симптомами) и васкулит или воспаление мелких кровеносных сосудов в периферическом нерве.
Обычно требуется невролог, чтобы установить диагноз, основанный на анамнезе и физическом осмотре. Всего тестов:
- электродиагностическое исследование, которое включает исследования нервной проводимости (NCS) и игольчатую электромиографию (EMG).
- Лабораторные исследования на антитела IgM GM1 также часто проводятся.
Как лечится MMN?
В настоящее время установлено, что внутривенный иммуноглобин (IVIg), препарат антител, полученный от здоровых добровольцев, может быть легко введен через вену руки и приносит пользу пациентам с MMN.Это только для лечения этого расстройства, одобренного Федеральным управлением по лекарственным средствам (FDA) и регулирующими органами в Европе и Канаде. IVIg может привести к улучшению двигательной функции у большинства пациентов с MMN, при этом ответ варьируется от минимального до очень большого. Раннее лечение вскоре после появления симптомов всегда более эффективно. Лечение обычно не полностью устраняет все симптомы, и тем пациентам, которые действительно реагируют, потребуются повторные сеансы лечения для поддержания их улучшения.Пациентам обычно требуется повторное лечение каждые 2-5 недель, а со временем могут потребоваться повышенные дозы ИГВВ.
IVIg не является лекарством от MMN, но в настоящее время никакая другая терапия не доказала свою эффективность. У ограниченного числа пациентов химиотерапевтический препарат против рака, циклофосфамид, временно эффективен. Однако его использование ограничено токсическими побочными эффектами и рисками, которые возникают при постоянном употреблении. Другие иммуносупрессивные методы лечения, такие как кортикостероиды и плазмаферез, неэффективны и фактически могут усугубить болезнь.
Многие исследователи по всему миру работают над тем, чтобы лучше понять MMN.
Ресурсы и обновления
Мультифокальная моторная нейропатия: противоречия и приоритеты
Введение
Несмотря на 30-летние исследования, все еще существуют значительные неизвестные и противоречивые данные, связанные с мультифокальной моторной невропатией (ММН), включая патофизиологию заболевания, диагностические критерии и лечение (таблица 1). Прежде всего, это связано с лежащей в основе патофизиологией, а именно с тем, представляет ли MMN аксональную или демиелинизирующую невропатию и сосредоточена ли основная патофизиология на узле Ранвье.В свою очередь, это обсуждение способствует рассмотрению терапевтических подходов, и этот вопрос становится все более актуальным в эту развивающуюся эпоху точной медицины. Известно, что MMN представляет собой хроническую прогрессирующую иммуноопосредованную моторную невропатию, клинически характеризующуюся прогрессирующей асимметричной слабостью, а электрофизиологически — частичной блокадой моторной проводимости. Хотя MMN можно считать редким с оценкой распространенности от 0,6 до 2 на 100 000 населения, 1 2, вероятно, будет недостаточно признанным субъектом.В дополнение к лучшему пониманию основных механизмов, в последнее время также появились разработки в области использования нервно-мышечного ультразвука и МРТ для помощи в диагностике MMN и в дальнейшем выяснении его патофизиологических механизмов. Таким образом, в настоящем Обзоре будет проведен критический анализ знаний, накопленных о MMN за последние 30 лет, что приведет к современному подходу к терапии.
Таблица 1Противоречия и приоритеты в мультифокальной моторной нейропатии (MMN)
Клинический фенотип
Классически MMN представляет собой асимметричную чисто моторную множественную мононевропатию верхних конечностей с часто выраженным истощением, несмотря на короткую историю слабости.MMN чаще поражает мужчин с соотношением мужчин и женщин 2,7: 1. Возраст появления симптомов может составлять от 20 до 70 лет, средний возраст — 40 лет.1 3 4 Первым симптомом чаще всего является слабость дистального отдела верхней конечности, но с относительной щадью сгибателей пальцев1. пациентов. У многих с начальным поражением нижних конечностей позже развиваются симптомы верхних конечностей. Чаще сообщается о поражении недоминантной верхней конечности. Фасцикуляции и судороги заметны и наблюдаются у 40% пациентов, а в некоторых случаях могут вызывать локальную гипертрофию мышц.Обострение слабости от холода является обычным явлением, о чем сообщалось в 83% случаев в одном исследовании5. Глубокие сухожильные рефлексы пораженной конечности обычно снижены, но также могут быть нормальными.
Наличие сенсорных симптомов, признаков или нейрофизиологических аномалий, особенно при постановке диагноза или на ранних стадиях заболевания, должно вызвать значительную диагностическую неопределенность и побудить к дальнейшим исследованиям, включая рассмотрение прицельной биопсии нерва. Однако в одной серии случаев у 22% было аномальное чувство вибрации в дистальном отделе нижней конечности, причем в этих случаях средняя продолжительность заболевания была больше, чем у тех, у которых не было сенсорных данных.1
Вовлечение за пределы конечностей встречается редко, с разрозненными сообщениями о поражении диафрагмальных и черепных нервов.6 Диагностические критерии MMN были предложены Европейской федерацией неврологических обществ и Обществом периферических нервов и опубликованы в 2010 году (во вставке 1 перечислены клинические критерии для MMN) .7
Вставка 1Клинические критерии мультифокальной моторной нейропатии, предложенные Европейской федерацией неврологических обществ7
Основные критерии — должны присутствовать оба
Медленно прогрессирующая или ступенчато-прогрессирующая, очаговая, асимметричная слабость конечностей, то есть моторное поражение в распределении двигательных нервов по крайней мере двух нервов в течение более 1 месяца.Если симптомы и признаки присутствуют в распределении только одного нерва, можно поставить диагноз
Объективных сенсорных отклонений нет, за исключением незначительных отклонений чувствительности к вибрации в нижних конечностях
Поддерживающие клинические критерии
Преобладающее поражение верхней конечности
Снижение или отсутствие сухожильных рефлексов пораженной конечности
Отсутствие поражения черепных нервов
Судороги и фасцикуляции пораженной конечности
Реагирование на ответ или мышечной силы на иммуномодулирующее лечение
Критерии исключения
Признаки верхнего двигательного нейрона
Заметное поражение бульбара
Сенсорное нарушение более выражено, чем незначительная потеря вибрации в нижних конечностях
Диффузная симметричная слабость в течение первых недель
Клиническая нейрофизиология 9019
Выявление моторного блока проводимости (CB) является ключевым нейрофизиологическим критерием в диагностике MMN (вставка 2).7 Два наиболее часто поражаемых нерва — это срединный и локтевой нервы 3, расположенные в сегментах предплечья, а не в типичных местах сдавления. CB, наблюдаемый при MMN, уникален тем, что он влияет на моторные волокна исключительно с нормальной сенсорной проводимостью через тот же сегмент смешанных нервов.2 3 Кроме того, блокада является очаговой и возникает внезапно и, по крайней мере, на самых ранних стадиях заболевания, двигательной проводимости. дистальнее места блока может оставаться нормальным.
Вставка 2Электрофизиологические критерии блокады проводимости7
1.Определенная блокада моторной проводимости (CB) *
Снижение площади отрицательного пикового комбинированного потенциала моторного действия (CMAP) при проксимальной и дистальной стимуляции не менее чем на 50% независимо от длины нервного сегмента (медиана, локтевая и малоберцовая). Амплитуда отрицательного пика CMAP при стимуляции дистальной части сегмента моторным CB должна составлять> 20% нижнего предела нормы и> 1 мВ, а увеличение длительности отрицательного пика CMAP от проксимального до дистального должно быть ≤30%
2. Вероятный моторный CB *
Уменьшение площади отрицательного пика CMAP не менее 30% на длинном сегменте нерва верхней конечности с увеличением длительности отрицательного пика CMAP от проксимального до дистального ≤30%
OR
Уменьшение площади отрицательного пика CMAP на не менее 50% (то же, что и определенное) с увеличением длительности CMAP от проксимального до дистального отрицательного пика> 30%
3.Нормальная проводимость сенсорных нервов в сегментах верхних конечностей с CB (см. Критерии исключения)
Copyright 2010 Peripheral Nerve Society. Используется с разрешения компании John Wiley and Sons.
* Признаки ХБ должны быть обнаружены в местах, отличных от обычных синдромов ущемления или сжатия.
Критерии для определенного CB были подробно обсуждены с различными критериями для разных нервов в зависимости от сложности получения согласованных сложных потенциалов двигательного действия (CMAPs) из соответствующих мест.2 3 8–10 Например, определение CB как определенного в большеберцовом нерве требует большего декремента CMAP, чем в сегменте предплечья срединного нерва. Есть свидетельства того, что декремент CMAP для обнаружения блока может быть более точно измерен по декременту площади CMAP, а не по амплитуде.9
Отмена фазы из-за временной дисперсии, приводящей к ложному ХБ, является частой находкой при демиелинизирующих невропатиях и может быть существенной проблемой при диагностике ХБ при MMN.9 В настоящее время не существует надежных и воспроизводимых методов оценки проксимальных участков ХБ.Стимуляцию моторных корешков иглой провести сложно из-за значительной вероятности ложноположительных результатов, и она плохо переносится. Многие критерии CB в MMN также включают условие о максимальной допустимой временной дисперсии. Точно так же очень трудно надежно определить CB, когда амплитуда вызванной дистальной CMAP меньше 1 мВ.
Электромиография (ЭМГ) почти всегда выявляет значительную хроническую денервацию и реиннервацию мышц, снабжаемых нервами с CB, демонстрируя, что дегенерация аксонов является важной характеристикой MMN даже с самого раннего начала заболевания.3
Были сообщения о MMN с типичными клиническими признаками, но без выявления CB.4 Возможная причина — очень проксимальные или дистальные места CB, где обычная электрофизиология не может обнаружить блок. Другая возможность заключается в том, что исследования проводимости проводились только в клинически пораженных конечностях, в то время как CB может быть обнаружен также в нервах, иннервирующих мышцы с нормальной силой. По-видимому, не наблюдается каких-либо существенных различий в клинических характеристиках и ответе на лечение между пациентами с фокальной блокадой и без нее.4
Дифференциальный диагноз
У пациента с симптомами слабости, указывающими на синдром нижнего двигательного нейрона (LMN), помимо MMN, следует учитывать другие диагнозы, включая боковой амиотрофический склероз (ALS) и, в частности, вариант LMN прогрессирующей мышечной атрофии (PMA), поздние формы спинальной мышечной атрофии (SMA), мономерная амиотрофия, включая болезнь Хираямы, фокальные и чисто моторные варианты хронической воспалительной демиелинизирующей полинейропатии (CIDP), мультифокальную приобретенную демиелинизирующую сенсорную и моторную невропатию (MADSAM), часто называемую Синдром Льюиса-Самнера, дистальные наследственные моторные невропатии и наследственная невропатия со склонностью к параличу от давления (HNPP).11 Вариант острой моторной аксональной нейропатии (AMAN) синдрома Гийена-Барре (GBS) рассматривается в чисто моторных проявлениях с острым началом. Сообщалось о случаях внезапного появления MMN, хотя это необычно.12 Мониторинг часто выявляет прогрессирование MMN после 1 месяца, тогда как AMAN имеет тенденцию демонстрировать монофазный курс с плато через 4 недели от появления симптомов с последующим периодом выздоровления.
MMN — важный диагноз, который следует учитывать при наличии хотя бы частично эффективного лечения.Некоторые особенности нетипичны для MMN, особенно наличие бульбарного или респираторного поражения или сенсорного поражения. Дифференциальный диагноз обычно можно отличить от MMN с помощью соответствующей визуализации шейного отдела спинного мозга (болезнь Хираямы), электрофизиологического (MADSAM, PMA), генетического (SMA, HNPP и часть dHMN), серологического (AMAN обычно ассоциируется с антителами IgG против GM1). и GD1a) и в некоторых случаях анализ спинномозговой жидкости (MADSAM, CIDP и варианты, GBS).11 Хотя в некоторых случаях диагноз все еще затруднен, и требуются серийные оценки и даже испытания терапии с соответствующим мониторингом.
МРТ и нервно-мышечное УЗИ
Поддерживающим критерием для диагностики MMN является обнаружение диффузного отека нервов плечевого сплетения с помощью МРТ.7 Ультразвук нервов также становится инструментом для выявления таких аномалий и возможных участков проксимального отдела CB.
МРТ корешков шейных нервов и плечевого сплетения может продемонстрировать аномалии корня шейки матки и гиперинтенсивность Т2 сплетения и увеличение MMN у 35–50% пациентов.13 14 Такие аномалии также можно увидеть при других воспалительных невропатиях, таких как CIDP. Отклонение от нормы МРТ не коррелировало с конкретными клиническими характеристиками или ответом на лечение у пациентов с MMN.
Ультразвук высокого разрешения (HRUS) — новый инструмент в оценке патологии периферических нервов. Мультифокальное увеличение нерва можно идентифицировать в плечевом сплетении, нервных корешках и периферических нервах, причем эти изменения проявляются в нервах как с клиническими и электрофизиологическими отклонениями, так и без них.15 Одно исследование показало значительно увеличенные площади поперечного сечения (CSA) в срединных, локтевых и большеберцовых нервах у пациентов с MMN по сравнению с контрольной группой.16 Внутри нервная изменчивость и асимметрия CSA наблюдались в срединном нерве пациентов с MMN, что свидетельствует о значительной очаговой активности. нервных аномалий.
Сравнительное исследование МРТ и HRUS корешков шейных нервов и плечевого сплетения у не получавших лечения пациентов с CIDP или MMN продемонстрировало результаты МРТ увеличения нервов и / или гиперинтенсивности T2 у 50% пациентов с MMN и результаты HRUS — увеличение нервов в 67%.14 Нарушения визуализации при любом способе лечения наблюдались у 79% пациентов с MMN, половина из этой группы имела как аномалии МРТ, так и HRUS, 36% — только аномалии HRUS и 14% — только результаты МРТ.
Ультразвуковые исследования показали многообещающие возможности дифференцировать MMN от ALS. Исследования двигательных периферических нервов, корешков шейных нервов и / или плечевого сплетения выявили повышенные показатели CSA в MMN по сравнению с пациентами с ALS.17-19 Также были предложены протоколы и модели ультразвукового исследования для дифференциации MMN и ALS с чувствительностью более 87%. и особенности, приближающиеся к отображению 100%.Помимо CSA периферического нерва, недавнее исследование с использованием ультразвука нервов показало, что соотношение дистально-проксимального отдела медианного и локтевого нервов может быть полезным для дифференциации БАС от MMN.20 Однако неясно, может ли HRUS различать MMN и моторные варианты CIDP.
Нарушения визуализации в MMN, как правило, мультифокальные и могут коррелировать с участками ХБ, но также могут быть идентифицированы в нервах, в которых не обнаружены клинические или электрофизиологические отклонения. и только электрофизиологическое обследование с аномалиями визуализации, потенциально обнаруживаемыми на более ранней стадии течения заболевания.Кроме того, диагностические протоколы, состоящие из обследования в определенных местах, могут быть полезны, как показано в исследованиях на основе HRUS18. 19 Оценка на основе HRUS, вероятно, будет более дешевым, безопасным и доступным методом, чем МРТ. Однако оба метода требуют опыта работы и интерпретации и требуют определения нормальных значений для популяции, чтобы они были частью объективной оценки.
Патофизиология
Считается, что MMN является иммуноопосредованной нейропатией. Патофизиологический механизм, лежащий в основе ХБ, изначально предполагался как следствие очаговой демиелинизации, но появляется все больше доказательств того, что функциональные нарушения в узле Ранвье могут быть причиной блокады.Термин « нодопаранодопатия » включает невропатии, вызванные дисфункцией узлов и параноузлов.22 Дисфункция шванновских клеток в паранодальной области может быть важным компонентом нодопаранодопатии с очень очаговой демиелинизацией в этой области, необходимой для открытия параноза для иммунной атаки. .
Исследования нервной возбудимости MMN показали параметры возбудимости, согласующиеся с гиперполяризацией, что также наблюдается в постишемических нервах, включая большую сверхвозбудимость аксонов дистальнее места CB.23 Деполяризация в месте ХБ может привести к этому наблюдению из-за внутриклеточного накопления натрия (Na + ), возможно, из-за натриевого канала или дисфункции Na + / калия (K + ) -насоса. Недавнее математическое моделирование, проведенное Гаргом и его коллегами, позволило предположить снижение функции ионных каналов Na + и K + вдоль аксона дистальнее CB.24 Эти функциональные нарушения аксональной мембраны, как предполагается, приводят к увеличению эктопической генерации, проявляющейся как положительная симптоматика. такие как фасцикуляции и судороги.23 Дальнейшее исследование, в котором поляризующие постоянные токи подавались в участки ХБ, как было определено с помощью инчинг-исследований, показало, что блоки могут быть деполяризующими, гиперполяризующими или и тем, и другим. применяется, с ожидаемым ухудшением гиперполяризации и деполяризацией для улучшения CB в участках гиперполяризации мембраны, с противоположным применением для участков деполяризации. Авторы предположили, что их результаты могут отражать эволюцию заболевания с течением времени с деполяризацией, предшествующей более позднему развитию гиперполяризационных блоков.
CB в MMN оказался зависимым от активности. Одно исследование показало временное снижение соотношения проксимально-дистального CMAP после максимального произвольного сокращения (MVC) 26. Также наблюдалось снижение силы, создаваемой пораженными мышцами после сокращения. Преходящий CB или временная дисперсия наблюдалась после MVC у шести пациентов с MMN без явного CB, выявленного при рутинных электродиагностических тестах.27 Другое исследование с участием 19 пациентов с MMN не смогло продемонстрировать зависимый от активности CB.28 Однако в нервных сегментах с ранее существовавшим CB, MVC привело к временной дисперсии.Помимо различий в методологии, разные результаты исследований могут отражать характер или степень лежащего в основе блока. Механизмом этих изменений, вероятно, является дисфункция узловых натриевых каналов, гиперактивность Na + / K + -насоса или другие узловые / паранодальные функциональные нарушения.
Восемьдесят три процента пациентов с MMN сообщают о холодовом парезе. 5 Это свидетельствует о выраженном функциональном нарушении, а не о очаговой демиелинизации, поскольку холод имеет тенденцию улучшать симптомы и ХБ, вторичный по отношению к демиелинизации.Было показано, что охлаждение нервов человека вызывает деполяризацию аксонов, вероятно, из-за снижения активности термочувствительного Na + / K + -насоса.29 В MMN деполяризующие CBs могут усугубляться через этот механизм с соответствующим обострением симптомов. Исследование, сравнивающее влияние охлаждения на параметры возбудимости моторных и сенсорных аксонов, обнаружило существенные различия между ними.30 В моторных аксонах охлаждение приводило к изменениям, согласующимся с деполяризацией аксонов, включая усиление порогового электротонуса (что определяется снижением порогового значения). изменение формы сигналов порогового электротонуса) и повышение крутизны зависимости вольт-амперной характеристики (I / V).30 31 Эти находки могут отражать различия в подтипах ионных каналов, выраженных на моторных и сенсорных аксонах, с их участием в MMN, возможно, объясняющим его чисто моторные проявления. Исследования нервной возбудимости и феномен холодового пареза показывают, что MMN нельзя объяснить только демиелинизацией, тем самым подтверждая концепцию нодопаранодопатии.
Анти-ганглиозидные антитела
Присутствие анти-GM1 IgM-антител было впервые описано в MMN в 1988 году.Эти антитела присутствуют по крайней мере в 40% случаев.1 3 32 Чувствительность низкая, и они обнаруживаются у части неврологически нормальных пациентов, пациентов с БАС и другими невропатиями. Однако в условиях высоких положительных титров и при подозрении на иммуноопосредованную моторную нейропатию специфичность может превышать 90% .32 В MMN эти антитела, вероятно, вырабатываются ограниченным числом клонов В-клеток.
Факторы, приводящие к развитию MMN, недостаточно изучены. Генетическая предрасположенность к аутоиммунным заболеваниям подтверждается повышенной частотой других аутоиммунных заболеваний, присутствующих у пациентов с MMN и их родственниками первой степени родства, а также более высокой частотой гаплотипа человеческого лейкоцитарного антигена DRB1 * 15, который также связан с рассеянным склерозом и CIDP. среди пациентов с MMN по сравнению с контрольной группой.34 35 Предыдущий инфекционный триггер может играть роль, потенциально за счет молекулярной мимикрии ганглиозидов или других эпитопов, хотя пока нет никаких четких доказательств, подтверждающих это. Небольшое исследование показало, что антитела против Campylobacter jejuni были более частыми, чем ожидалось, у пациентов с MMN, но это не было подтверждено более поздним исследованием36. 37 Моноклональная гаммапатия IgM была выявлена у 7% пациентов с MMN по сравнению с 2% здоровых людей из контрольной группы. .38 Значение этого открытия неясно и может указывать на аутоиммунитет В-клеток или сопутствующее лимфопролиферативное заболевание, которое может быть или не быть патогенетически связанным.
Ганглиозиды представляют собой гликосфинголипиды, присоединенные к фрагментам сиаловой кислоты. GM1 повсеместно присутствует в большом количестве и обнаруживается в центральной и периферической нервной системе. 39 В периферических нервах он обнаруживается в аксолемме и миелине, преимущественно в узловых и паранодальных областях и концентрируется в обогащенных холестерином доменах плазматических мембран. Ганглиозиды выполняют несколько функций, включая поддержание плотных контактов, кластеризацию ионных каналов с поддержанием калиевых каналов в паранодальных / межузловых областях и натриевых каналов в узлах, а также клеточный гомеостаз кальция.39–41
Исследования показали, что антитела против GM1 являются патогенными как по прямым, так и по непрямым механизмам (рис. 1). Антитела против GM1 могут вызывать прямое функциональное нарушение за счет увеличения калиевого тока в параузлах и нарушения кальциевых сигнальных путей.41, 42 Они также могут вызывать непрямую нервную дисфункцию через классический путь комплемента.43 В исследованиях на животных активация комплемента и мембранная атака комплексны. образование приводит к разрушению кластеров узловых натриевых каналов и кластеров паранодальных калиевых каналов.42 44 Эти изменения могут привести к подавлению натриевого тока и утечке управляющего тока. Отложение и активация комплемента предотвращалось добавлением иммуноглобулинов.43 Модель MMN, полученная на основе индуцированных плюрипотентными стволовыми клетками человека (IPSC), показала как опосредованное антителами комплемент-зависимое, так и независимое от комплемента нарушение гомеостаза кальция мотонейронов и повреждение аксонов.41 Нарушения были уменьшены с применением иммуноглобулина. Хотя открытие этой модели MMN у человека in vitro дает новые идеи, их следует интерпретировать с учетом ее ограничений, которые включают отсутствие глиальных клеток и, в частности, шванновских клеток и относительную незрелость изучаемых нейронов.
Рисунок 1Упрощенная схематическая диаграмма миелинизированного аксона моторного нерва узловой / паранодальной области и потенциальных механизмов дисфункции и повреждения нерва при мультифокальной моторной невропатии. IgM к GM1 может вызывать как прямые, так и косвенные (через комплемент) нарушения мотонейронов и, следовательно, приводить к блокаде фокальной проводимости (A). Прямые эффекты включают нарушение гомеостаза кальция с увеличением внутриклеточной концентрации кальция и увеличением тока калия (B). Косвенные эффекты могут быть опосредованы классическим путем комплемента с образованием комплекса мембранной атаки (MAC) (C).Это может привести к разрушению кластеров узловых натриевых каналов, разрушению параузлов с измененными кластерами калиевых каналов и повреждению нейронов с окончательной дегенерацией аксонов. NaV, потенциал-закрытый натриевый канал; Kv, калиевый канал, управляемый напряжением; I Na , натриевый ток; I K , ток калий.
Используя непрямую иммунофлуоресценцию на нервных волокнах крыс, инкубированных с сыворотками 11 пациентов с MMN, Гарг и др. идентифицировали только одного пациента с паранодальным окрашиванием, несмотря на то, что 55% пациентов были положительными по анти-GM1 IgM.24 Напротив, 9 из 11 пациентов с ХВДП с ХБ показали окрашивание в узле, параузле и / или миелине. Это подтверждает мнение о том, что MMN и CIDP имеют разные механизмы заболевания и антигены-мишени. Хотя это исследование не является поддерживающим, оно не исключает MMN как возможную нодопаранодопатию. Различия между человеческим и крысиным эпитопом могут ограничивать связывание антител, а внутривенный иммуноглобулин (ВВИГ), вводимый в качестве лечения, также может влиять на результаты.
Фокальность аномалий вызывает вопросы относительно патофизиологических механизмов, приводящих к такой избирательности.Было показано, что сыворотка пациентов с MMN нарушает гемато-нервный барьер 45, при этом двигательные нервы более восприимчивы, чем сенсорные нервы. Нет существенной разницы в количестве GM1, обнаруженного на двигательных или сенсорных нервах, но различия в составе церамидных фрагментов ганглиозидов могут модулировать аффинность связывания антител и восприимчивость к опосредованному антителами повреждению двигательных и сенсорных нервов.46 GM1 играет решающую роль в поддержание целостности двигательных нервов, но не так существенно для сенсорных нервов.40 Инкубация сенсорных нейронов, полученных из IPSC человека, с сыворотками пациентов с MMN привела к связыванию антител против GM1 с сенсорными нейритами. 41 Однако сенсорные нейриты не подвергаются повреждению, как это наблюдается в двигательных нейритах. Это говорит о том, что двигательные нейроны более уязвимы к повреждению, опосредованному антиганглиозидными антителами, чем сенсорные нейроны. Возможное объяснение — наличие кофакторов на двигательных нейронах, но отсутствует на сенсорных нейронах, играющих критическую роль в разоблачении эпитопов и облегчении узловой дисфункции и повреждения нервов.41 год
Другие антитела и антигены-мишени
В модели, полученной из IPSC человека, Harschnitz и др. обнаружили, что было значительно повышенное связывание IgM с нейритами после инкубации с сыворотками пациентов, положительными или отрицательными по MMN анти-GM1, по сравнению с контрольными сыворотками. 41 Целью антител обеих групп сывороток оказался GM1. Это открытие, вероятно, частично связано с техническими факторами, ограничивающими чувствительность обнаружения антител против GM1. Несколько исследований показали повышенный уровень обнаружения анти-GM1 IgM в сочетании с гликолипидным галактоцереброзидом (GalC) с повышенной чувствительностью до 81%, но с небольшим снижением специфичности.47 48 Добавление GalC, вероятно, усиливает связывание антител, возможно, за счет лучшего воздействия эпитопов GM1.
Другая возможность в случаях отрицательных антител к GM1 — это наличие антител, направленных против других антигенов. Несколько исследований были направлены на их выявление. Узловые белки глиомедин и нейрофасцин-186 (NF186) были предложены в качестве потенциальных мишеней, учитывая их роль в кластеризации узловых натриевых каналов. Notturno et al показали, что 62% пациентов с MMN имели антитела к глиомедину или NF186 с использованием синтетических крысиных пептидов.Однако это не было подтверждено анализами клеточного связывания с использованием трансфецированных клеток.49 Два исследования, в которых дополнительно изучали антитела против паранодальных белков контактина-1 и нейрофасцина-155 с использованием человеческих белков, не обнаружили антител против них ни у одного пациента с MMN.50 51 Вероятная причина Эти противоположные результаты заключаются в использовании различных белков в их анализах, крыс в первом и человека в двух последующих исследованиях. Pestronk и др. предположили, что антитела против гепарин-дисахарида NS6S связаны с приобретенными хроническими моторными невропатиями.52 Они присутствовали у 43% пациентов с моторной нейропатией и 57% с MMN. Nobile-Orazio et al идентифицировали 23% пациентов с MMN с антителами IgM к NS6S, но это не было статистически значимым по сравнению с контрольной группой.47 Таким образом, диагностическая и патогенная значимость антител против NS6S остается неопределенной.
Патологические изменения
Было проведено немного исследований, изучающих патологические изменения пораженных нервов. Каджи и его коллеги описали пациента, у которого для патологического обследования удалили медиальный грудной нерв, который находился дистальнее места CB в плечевом сплетении.21 Демиелинизирующие особенности аксонов большого диаметра, которые были тонко миелинизированы или лишены миелина, и маленькие луковицы лука наблюдались при пониженной плотности волокон. Инфильтратов воспалительных клеток не наблюдалось. Auer и соавт. сообщили о случае синдрома нижних мотонейронов, указывающего на MMN, с электрофизиологическими данными, соответствующими демиелинизации плечевых сплетений, но без выявления CB.53 Биопсия проксимального правого локтевого нерва продемонстрировала признаки, соответствующие хронической демиелинизации луковицами лука и отсутствие воспалительных клеток.
Напротив, Тейлор и его коллеги идентифицировали дегенерацию и потерю мультифокальных волокон (особенно крупных волокон) и значительное количество регенеративных кластеров в биоптатах фасцикулярного нерва, взятых на участках CB в восьми нервах от семи пациентов с MMN. демиелинизация, включая отсутствие образования луковиц. Небольшие периваскулярные лимфоцитарные инфильтраты были обнаружены в двух нервах. Авторы предположили, что MMN была аксонопатией без демиелинизации, а CB — это опосредованная антителами каналопатия, сосредоточенная в узлах Ранвье.
Эти два несопоставимых открытия, одно о хронической демиелинизации, а другое о мультифокальной дегенерации и регенерации волокон, открывают несколько возможностей. Одна из возможностей состоит в том, что различные патологические изменения происходят на разных участках пораженных нервов, с хроническими демиелинизирующими изменениями на участках, удаленных от CB, возможно, вторичными по отношению к первичному поражению, вызывающему CB. Другая возможность состоит в том, что различные лежащие в основе патологические механизмы приводят к различным патологическим изменениям. Фокальные и моторные варианты CIDP могут иметь клиническую картину, очень похожую на MMN, а также демонстрировать моторный CB при электрофизиологических исследованиях.Однако можно ожидать, что патология нервов в фокально-моторном ХВДП продемонстрирует демиелинизацию как характерную черту (рис. 2).
Рисунок 2Биопсия проксимального фасцикулярного нерва, взятая из смешанных моторных и сенсорных нервов. В обоих случаях преобладали фокально-моторные нейропатии с некоторыми аномалиями визуализации, которые позволили выбрать место для биопсии нерва. В случае А (А1 – А4) была очаговая гипертрофическая демиелинизация, поэтому был поставлен диагноз очаговая хроническая воспалительная демиелинизирующая полинейропатия, тогда как в случае B (B1 – B3) наблюдались легкие нейрогенные изменения без признаков воспалительной демиелинизации.Биопсия фацикулярного нерва из левого плечевого сплетения (а). (A1) Препараты с раздвоенными волокнами, показывающие области демиелинизации между стрелками. (A2) Полупрозрачный эпоксидный срез, окрашенный метиленовым синим, показывает очевидную очаговую потерю миелинизированных волокон. (A3) Электронная микроскопия области потери волокна, показывающая частые небольшие образования луковиц (стрелки). (A4) Электронная микроскопия при более высоком увеличении показывает небольшую луковицу без волокна в центре. Биопсия фацикулярного нерва от левого срединного нерва в месте блокады проводимости (B).(B1) Препараты с раздвоенными волокнами, показывающие нормальные миелинизированные волокна без демиелинизации. (B2) Полупрозрачный эпоксидный срез, окрашенный метиленовым синим, показывает нормальную плотность миелинизированных волокон. (B3) На более высокой мощности стрелки, показывающие регенерирующий кластер (стрелки), но не луковицы. (С любезного разрешения П. Джеймс Б. Дайк и Джанин К. Энгельстад).
Что касается патологии сенсорных нервов, исследование биопсий сенсорных нервов у 11 пациентов обнаружило очень незначительные демиелинизирующие особенности.55 Эти данные предполагают, что сенсорные нервы также могут подвергаться поражению, хотя и гораздо меньшей степени тяжести, чем двигательные нервы.Однако, учитывая отсутствие сенсорных симптомов и электрофизиологических аномалий в MMN, а также в случаях биопсии и наблюдаемых очень незначительных патологических изменениях, трудно сделать какие-либо убедительные выводы, хотя сенсорное вовлечение в позднюю стадию заболевания было зарегистрировано в значительном количестве. дел.1 3
Лечение
Учитывая вероятную иммунопатогенную основу, иммуномодуляторы широко используются в лечении MMN. ВВИГ в настоящее время является препаратом первой линии, эффективность которого подтверждается данными нескольких рандомизированных контролируемых исследований (РКИ).Однако данные РКИ в поддержку неиммуноглобулиновых препаратов отсутствуют.
Внутривенный иммуноглобулин
Благоприятный эффект ВВИГ в отношении увеличения мышечной силы был впервые описан в 1992 году. На сегодняшний день было проведено пять РКИ по применению ВВИГ при ММН, и все они продемонстрировали явное преимущество. Четыре из них были частью Кокрановского метаанализа, который продемонстрировал улучшение мышечной силы у 78% пациентов, получавших ВВИГ, по сравнению только с 4%, получавшими плацебо. У 39% пациентов, получавших ВВИГ, наблюдалось улучшение инвалидности, но это было заметно. не является статистически значимым, что, вероятно, связано с отсутствием хорошей шкалы инвалидности, подходящей для необычного распределения слабости в MMN.Последнее РКИ представляло собой двойное слепое перекрестное исследование с 44 пациентами, рандомизированными в две группы, одна из которых получала 12 недель ВВИГ, затем 12 недель плацебо, а другая получала такое же лечение в обратном порядке.57 Средняя максимальная сила захвата пораженной руки. увеличился на 3,75% с IVIG и снизился на 31,38% с плацебо. Ухудшение трудоспособности наблюдалось у 35,7% субъектов, получавших плацебо, но не после перехода на ВВИГ. Это сравнивалось с 11,9%, чья инвалидность ухудшалась во время приема ВВИГ, но не во время периода плацебо.Шестьдесят девять процентов субъектов были преждевременно переведены с плацебо на открытые внутривенные иммуноглобулины из-за значительного функционального ухудшения на плацебо. Авторы пришли к выводу, что лечение ВВИГ в MMN явно улучшило мышечную силу и инвалидность.
В долгосрочной перспективе большинству пациентов требуется поддерживающая ВВИГ для предотвращения клинического ухудшения. Несмотря на продолжающийся внутривенный иммуноглобулин, у значительной части пациентов наблюдается прогрессирование заболевания со снижением мышечной силы, но все же лучше, чем до лечения.1 8 58–60 Доза ВВИГ, необходимая для обслуживания, увеличивается со временем. Серийная электрофизиологическая оценка продемонстрировала доказательства реиннервации и демиелинизации / потери аксонов, хотя ХБ может сохраняться, несмотря на клиническое улучшение60. ВВИГ, хотя и полезен, способен лишь частично контролировать процесс заболевания и замедлять потерю аксонов.
Кошки и др. показали, что более тяжелая инвалидность или слабость были связаны с потерей аксонов и годами отсутствия лечения.1 Потеря аксонов была наиболее значимой детерминантой мышечной слабости, за которой следовали годы без лечения в другом исследовании.61 Исследование с использованием более высоких поддерживающих доз ВВИГ показало, что ВВИГ может значительно уменьшить дегенерацию аксонов, а также способствовать реиннервации.62 Таким образом, ранняя диагностика и лечебное учреждение имеют первостепенное значение для минимизации потери аксонов и увеличения инвалидности.
Подкожный иммуноглобулин
Подкожный иммуноглобулин (SCIG) имеет потенциальные преимущества перед ВВИГ, включая удобство для пациента, избежание госпитализации, потенциальную экономическую выгоду и лучший общий профиль побочных эффектов.SCIG также приводит к более стабильному уровню IgG в сыворотке, таким образом предотвращая нежелательные явления из-за нефизиологически высоких уровней IgG в сыворотке и колебания симптомов из-за эффекта конца дозы с IVIG.
В нескольких исследованиях описана терапия SCIG у пациентов, которые ранее получали поддерживающую ВВИГ, с использованием эквивалентных общих месячных доз. Они показали аналогичную эффективность между SCIG и IVIG.63 Метаанализ, изучающий эффективность и безопасность SCIG по сравнению с IVIG при MMN и CIDP, не показал значительных различий в силе мышц и снижение относительного риска умеренных или системных побочных эффектов на 28%.64 Двухлетнее последующее исследование показало, что SCIG эффективен для поддержания клинической стабильности.65
В открытом исследовании SCIG с 15 пациентами MMN использовалось соотношение доз 1,53: 1 SCIG-IVIG у пациентов, получавших менее 2 г / кг IVIG в месяц, и дозировка 1: 1 для пациентов, принимавших 2 г. /kg/month.66 У трех из шести пациентов, получавших эквивалентную дозу SCIG, ухудшилась сила, в то время как у всех пациентов, получавших дозу SCIG 1,53: 1, оставалась стабильной. Авторы предложили начинать дозирование SCIG в соотношении более 1: 1.Это контрастирует с исследованиями, демонстрирующими стабильность при эквивалентном дозировании.
Циклофосфамид
Имеются сообщения 1990-х годов, которые продемонстрировали клиническую пользу при пероральном или внутривенном введении циклофосфамида, в том числе в случаях, невосприимчивых к ВВИГ.67 Однако есть также описания случаев, в которых эффективность циклофосфамида сомнительна или неэффективна. все.59
В исследовании шести пациентов с MMN, получающих поддерживающую терапию ВВИГ, был добавлен пероральный циклофосфамид.67 Все шесть пациентов показали улучшение мышечной силы, функциональных нарушений и модифицированных оценок инвалидности Ранкина. Им удалось увеличить интервал между дозами ВВИГ, и трем пациентам удалось прекратить лечение на срок до 2 лет до возвращения симптомов, которые отреагировали на ВВИГ. Не сообщалось о более длительном наблюдении за этими пациентами.
Циклофосфамид имеет потенциально значительные побочные эффекты, включая миелосупрессию, геморрагический цистит, бесплодие, злокачественные новообразования и оппортунистические инфекции.67 Риск увеличивается при повторном лечении, которое обычно необходимо при MMN. У пациента, который невосприимчив к ВВИГ со значительным ухудшением слабости и / или функционального воздействия, может быть испытано лечение циклофосфамидом, но его следует рассматривать в контексте его профиля риска. Полное информированное согласие и тщательный мониторинг важны. Важно отметить, что существует мало доказательств того, что циклофосфамид или какой-либо другой вид терапии обратят установленную инвалидность, и поэтому в этой ситуации следует избегать использования потенциально вредных методов лечения.Учитывая неоднозначные отчеты о пользе, рандомизированное контролируемое исследование было бы очень полезным для выяснения эффективности циклофосфамида, а также для выяснения идеального пути введения и режима дозирования. Однако разработка и применение новых методов лечения сделали такое испытание маловероятным. Следовательно, циклофосфамид в качестве лечения MMN следует рассматривать как крайнюю меру с полным информированным согласием у тех, кто прогрессирует, несмотря на передовую практику лечения с помощью IVIG, и чьей независимости в значительной степени угрожает прогрессирующая MMN.
Стероиды и плазмаферез
Эффективность кортикостероидов при MMN была неутешительной, без пользы и в некоторых случаях клиническое ухудшение.59 Аналогичная ситуация наблюдалась с плазмообменом с недостаточной эффективностью, а также с некоторыми пациентами, показывающими ухудшение68. Причина этих неожиданных реакций на стероиды и плазмаферез неизвестна, но может быть связано с изменением баланса между повреждающими и регулирующими иммунными компонентами, что приводит к ухудшению клинического состояния.
Ритуксимаб
Ритуксимаб представляет собой химерное моноклональное антитело мыши и человека против CD20, которое приводит к истощению В-клеток. В нескольких небольших обсервационных исследованиях описана польза ритуксимаба, заключающаяся в улучшении мышечной силы и увеличении интервала между дозами внутривенного иммуноглобулина у пациентов, получающих поддерживающую терапию. .70 Открытое испытание шести пациентов, получавших поддерживающую терапию ВВИГ с ритуксимабом и наблюдаемых в течение 12 месяцев, не продемонстрировало снижения количества вводимых ВВИГ или изменения силы захвата, общей оценки Совета по медицинским исследованиям или оценки инвалидности.71 Необходимы дальнейшие исследования ритуксимаба и других моноклональных антител к CD20, чтобы определить, есть ли какие-либо преимущества. В настоящее время использование ритуксимаба при MMN не может поддерживаться вне клинических испытаний или, возможно, в качестве терапии спасения, когда другие методы лечения не дали результатов, с низким уровнем ожидания успеха.
Экулизумаб
Экулизумаб представляет собой гуманизированное моноклональное антитело против компонента С5 комплемента, которое действует, ингибируя терминальный путь активации комплемента. Учитывая, что активация комплемента, по-видимому, участвует в патофизиологии MMN, можно ожидать, что ингибирование предотвратит повреждение нервов.Доказано, что экулизумаб полезен при пароксизмальной ночной гемоглобинурии, заболевании, в патогенезе которого участвует активация комплемента. В открытом испытании экулизумаба в 13 случаях MMN были замечены улучшения в самооценке функциональной шкалы оценки и мышечной силы, измеренной с помощью миометрии, а также небольшое, но статистически значимое снижение среднего процента CB во всех изученных нервах. .72 Никаких изменений в интервале дозирования ВВИГ у тех, кто находился на поддерживающем лечении, не наблюдалось.Это краткосрочное неслепое исследование демонстрирует потенциальную клиническую пользу экулизумаба, но это необходимо установить в более крупных РКИ с более длительным периодом наблюдения.
Другие иммуномодулирующие препараты
Имеются сообщения об использовании других иммуномодулирующих средств, включая азатиоприн и бета-интерферон, с переменным эффектом.8 73 Дополнительное использование микофенолата оценивалось в рандомизированном контролируемом исследовании с 28 пациентами74. или доза ВВИГ. Таким образом, на данном этапе нет убедительных доказательств в пользу лечения этими агентами.
Выводы по лечению
В настоящее время единственным методом лечения, который однозначно доказал свою эффективность при лечении MMN, является ВВИГ / ПСИГ. Однако нет четкого консенсуса в отношении режимов лечения и того, когда следует сокращать, прекращать или начинать терапию. Многие случаи MMN могут демонстрировать стабильность в течение многих лет без терапии, и лечение может рассматриваться как поддержание их стабильности. Поэтому важно понимать траекторию состояния человека. Регулярный обзор с исследованиями нервной проводимости и слепая оценка функции периферических нервов, такая как оценка нарушения невропатии (NIS) или подшкала слабости NIS, могут значительно помочь в принятии решений о лечении.3 75 Не было показано никакого лечения, чтобы обратить вспять установленную атрофию. Траектория ухудшения при оценке является показанием к лечению, и оно должно состоять из ВВИГ или ПСИГ с использованием схем, указанных в клинических испытаниях. Если наблюдается улучшение или стабилизация траектории ухудшения, следует искать поддерживающую дозу ВВИГ / ПСИГ, которая поддерживает это улучшение. Практические рекомендации, предложенные Европейской федерацией неврологических обществ и Обществом периферических нервов, предполагают индукционную дозу ВВИГ 2 г / кг в течение 2–5 дней и поддерживающую дозу ВВИГ 1 г / кг каждые 2–4 недели или 2 г / кг каждые 1 день. до 2 месяцев в зависимости от реакции на лечение.7 IVIG / SCIG дорого обходится, и во многих странах их не хватает, поэтому необходимо приложить все усилия, чтобы ограничить количество, используемое для лечения MMN, как при всех состояниях. Следует разработать четкие инструкции по прекращению или сокращению терапии на основе испытаний отлучения от груди или прекращения терапии.
Другие иммунодепрессанты / иммуномодуляторы (циклофосфамид, ритуксимаб или экулизумаб) не показали воспроизводимого изменения естественного течения MMN в клинических испытаниях. Желательно, чтобы эти методы лечения проводились в рамках клинических испытаний.Однако, насколько нам известно, из-за редкости MMN они в настоящее время не ведутся. Полностью осознанное согласие и тщательный объективный мониторинг очень важны.
Моторная нейропатия — обзор
МНОГОФОКАЛЬНАЯ МОТОРНАЯ НЕЙРОПАТИЯ С БЛОКОМ ПРОВОДИМОСТИ
ПАТОГЕНЕЗ И ПАТОФИЗИОЛОГИЯ
Мультифокальная моторная нейропатия (ММН) с блокадой проводимости, иногда называемая моторной нейропатией с мультифокальным нарушением проводимости асимметричной двигательной слабостью и особенностями EDX приобретенных поражений SD, поражающих один или несколько локализованных сегментов двигательных нервных волокон. 67 Его патогенез неизвестен, но иммуно-опосредованный механизм поддерживается патологическими особенностями воспалительных демиелинизирующих изменений, благоприятным клиническим ответом на иммунодепрессанты и иммуномодулирующую терапию, а также многими поразительными сходствами с CIDP. 67 Патологические признаки включают демиелинизацию, иногда с признаками ремиелинизации в виде образования луковиц лука, и периваскулярные воспалительные инфильтраты, в основном лимфоцитов. Эти изменения наблюдались в черепных нервах, а также в проксимальных и дистальных сегментах периферических нервов, исследованных при вскрытии. 68 Хотя вовлечение моторных волокон отвечает за большинство клинических проявлений и особенностей EDX, аналогичные патологические изменения демиелинизации с воспалением также были зарегистрированы в сенсорных нервах, включая образцы икроножного нерва, исследованные при биопсии нерва. Отражая это, иногда наблюдаются сенсорные аномалии NCS.
Антитела к GM 1 ганглиозид были зарегистрированы у большого процента пациентов с MMN. 67 Этот фактор привел к мысли, что эти антитела играют патогенетическую роль в этом заболевании.Однако прямых доказательств в поддержку этого предложения нет. 69 Кроме того, наличие антител против GM 1 относительно неспецифично, поскольку они могут быть обнаружены при широком спектре других нервно-мышечных расстройств. Очень высокие титры антитела GM 1 , по-видимому, обладают большей специфичностью в отношении синдромов нижних мотонейронов, включая MMN. 70 Тем не менее, пациенты с MMN, у которых нет антител к GM 1 , по-прежнему реагируют на иммуномодулирующую терапию.Эта область быстро развивается. Возможно, что присутствие антител к GM 1 и другим антигенам может в конечном итоге оказаться более полезным при диагностике синдромов нижних мотонейронов, включая MMN. 70
ЭПИДЕМИОЛОГИЯ И ФАКТОРЫ РИСКА
MMN — редкое заболевание. Возраст начала охватывает многие десятилетия, от подросткового возраста до позднего взрослого возраста, но заболевание, как правило, начинается у молодых людей. Есть небольшое преобладание мужчин. Нет данных о предрасположенности к этому заболеванию у женщин во время беременности или к ухудшению во время беременности или в послеродовой период.
КЛИНИЧЕСКИЕ ОСОБЕННОСТИ И СВЯЗАННЫЕ ЗАБОЛЕВАНИЯ
MMN обычно является прогрессирующим заболеванием, которое вызывает мультифокальную и асимметричную слабость и иногда мышечную атрофию, сопровождающуюся судорогами, а иногда и фасцикуляциями. 67 Это заболевание часто начинается в верхних конечностях, и хотя оно может распространяться на нижние конечности, оно может оставаться локализованным или более выраженным в верхних конечностях на протяжении всего своего течения. В то время как некоторые ослабленные мышцы могут быть атрофическими, другие задействованные мышцы могут иметь нормальный объем, несмотря на хроническую слабость, что является клиническим признаком, указывающим на то, что блокада проводимости SD является механизмом слабости, а не потери аксонов.Поражение черепных нервов встречается редко, хотя были отмечены атрофия и слабость языка, а также другие бульбарные симптомы и признаки. Несмотря на то, что состояние преимущественно влияет на двигательную функцию, большинство пациентов в конечном итоге будут испытывать сенсорные симптомы, включая чувство онемения или покалывания, часто в отсутствие объективной сенсорной потери. Сенсорные симптомы и признаки чаще всего встречаются у пациентов с длительно сохраняющейся MMN. DTR часто отсутствуют или подавлены в распределении пораженных нервов, но, что удивительно, иногда они сохраняются.
MMN обычно следует очень хроническому вялотекущему течению, которое может длиться много лет. Иногда заболевание проявляется эпизодически, у пациентов наблюдается стабильное непрогрессивное течение, которое длится много месяцев или лет до того, как разовьется новый эпизод. С другой стороны, у некоторых пациентов есть более подострое течение, которое может привести к довольно выраженной широко распространенной слабости и инвалидности в течение нескольких месяцев. Сообщалось о редких случаях поражения ЦНС у пациентов с типичной НМН. Однако эти клинические признаки обычно скромны и редко напоминают комбинированные признаки заболевания верхних и нижних мотонейронов, наблюдаемые при боковом амиотрофическом склерозе (БАС). 67
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Многие клинические характеристики и особенности EDX MMN имеют близкое сходство с CIDP и родственными расстройствами. Фактически, MMN рассматривается некоторыми как вариант CIDP. 67 Таким образом, пациенты с преимущественно двигательной слабостью, сопровождающейся особенностями EDX приобретенного SD, должны быть обследованы на предмет тех же состояний, которые включены в дифференциальный диагноз CIDP. Как отмечалось ранее, MMN может иногда напоминать MADSAM. Однако важно отличать MMN от MADSAM, поскольку последнее условие больше напоминает типичный CIDP.В отличие от MMN, при MADSAM сенсорные симптомы и признаки могут быть замечены на ранней стадии, уровень белка в спинномозговой жидкости часто повышен, антитела против GM -1 отсутствуют, и пациенты могут реагировать на все типичные методы лечения, которые, как известно, эффективны при CIDP. . 45 Кроме того, некоторые пациенты с MMN могут внешне напоминать пациентов с заболеванием двигательных нейронов или БАС. Обычно MMN можно отличить от этих нарушений по более хроническому и вялотекущему течению, отсутствию симптомов и признаков верхних мотонейронов и, что наиболее важно, наличию приобретенных характеристик SD при тестировании EDX.Аксональная форма MMN была описана у девяти пациентов с симптомами и признаками, типичными для MMN, включая антитела против GM -1 , но без блокады проводимости или других признаков демиелинизации. 71 Некоторые из этих пациентов ответили на терапию ВВИГ. Неясно, представляют ли эти пациенты истинный аксональный вариант MMN или просто один из аспектов спектра этого заболевания. 72
ОЦЕНКА
Поскольку MMN может представлять собой вариант CIDP, важно оценивать пациентов с MMN так же, как пациентов с возможным CIDP.Следует рассмотреть возможность тщательного поиска M-белка, обследования костей скелета на предмет остеосклеротических или остеолитических поражений и тестирования на ВИЧ у пациентов из группы риска у пациентов с клиническими признаками, предполагающими MMN. ЦСЖ обычно нормален во всех аспектах, включая содержание в нем белка, что означает склонность MMN к поражению периферических нервов, а не корешков спинномозговых нервов. У большинства пациентов с MMN будут высокие титры антител против GM 1 . Как отмечалось ранее, присутствие антитела против GM 1 не является специфическим для MMN и не требуется для диагностики.Характеристики EDX этого расстройства обеспечивают его лабораторное подтверждение: они выявляют признаки мультифокального блока проводимости, обычно ограниченного двигательными волокнами (см. Рис. 49-2). Обычно участки поражения не являются обычными местами для защемления или компрессионной невропатии; например, очаговая блокада проводимости может быть обнаружена вдоль локтевого нерва предплечья, а не локтя.
ВЕДЕНИЕ
Поскольку MMN является редким заболеванием, данные об эффективности различных доступных методов лечения ограничены.Отдельные сообщения свидетельствуют о том, что преднизон, азатиоприн, микофенолятмофетил, ритуксимаб и плазмаферез редко бывают эффективными. 72 Однако могут быть полезны как ВВИГ, так и высокие дозы внутривенного циклофосфамида. 67,73 Данные рандомизированного двойного слепого плацебо-контролируемого исследования подтвердили эффективность ВВИГ при MMN. 74,75 Очевидно, что ВВИГ является более предпочтительным лечением, чем внутривенное введение циклофосфамида, из-за большего риска и осложнений, связанных с последним препаратом.Аналогично CIDP, IVIG можно вводить периодически для поддержания клинического улучшения. Следует подчеркнуть, что лечение MMN должно быть индивидуальным. Например, многие пациенты с MMN имеют очень скромные клинические симптомы, которые сохраняются без изменений в течение многих лет. Подходящим лечением таких пациентов может быть только наблюдение, а не риск и расходы на лечение ВВИГ или циклофосфамида.
ПРОГНОЗ И БУДУЩИЕ ПЕРСПЕКТИВЫ
Редкость этого заболевания и его относительно недавнее первоначальное описание в 1982 году являются факторами, ограничивающими опыт изучения его естественного происхождения.Тем не менее, у большинства пациентов с MMN, по-видимому, наблюдается хроническое, медленно прогрессирующее или стабильное течение в течение многих лет. С появлением внутривенного иммуноглобулина (ВВИГ), а также внутривенного введения циклофосфамида, вполне вероятно, что у многих пациентов заболевание может стабилизироваться, что приведет к умеренной инвалидности, если таковая имеется.
Ключевые фундаментальные проблемы, которые необходимо решить в отношении MMN, включают его точную патофизиологию, включая роль антител против GM 1 и их связь с CIDP. С клинической точки зрения требуется больше опыта, чтобы лучше сформулировать оптимальные терапевтические вмешательства.
Мультифокальная моторная нейропатия с блоками проведения: история вопроса, патофизиология, эпидемиология
Тейлор Б.В., Райт Р.А., Харпер С.М., Дайк П.Дж. Естественный анамнез 46 пациентов с мультифокальной моторной нейропатией с блокадой проводимости. Мышечный нерв . 2000 июня, 23 (6): 900-8. [Медлайн].
Slee M, Selvan A, Donaghy M. Мультифокальная моторная невропатия: спектр диагностики и ответ на лечение. Неврология . 2007 окт 23.69 (17): 1680-7. [Медлайн].
Руководство Европейской федерации неврологических обществ / Общества периферических нервов по лечению мультифокальной моторной нейропатии. Отчет совместной рабочей группы Европейской федерации неврологических обществ и Общества периферических нервов — первая редакция. J Peripher Nerv Syst . 2010 декабря 15 (4): 295-301. [Медлайн].
Cocito D, Bergamasco B, Tavella A, Poglio F, Paolasso I, Costa P и др.Мультифокальная моторная нейропатия на фоне лечения инфликсимабом. J Peripher Nerv Syst . 2005 декабрь 10 (4): 386-7. [Медлайн].
Harschnitz O, van den Berg LH, Johansen LE, Jansen MD, Kling S, Vieira de Sá R, et al. Патогенность аутоантител в модели, полученной из плюрипотентных стволовых клеток, индуцированной мультифокальной моторной невропатией. Энн Нейрол . 2016 Июль 80 (1): 71-88. [Медлайн].
Сусуки К., Расбанд М.Н., Тохьяма К., Койбути К., Окамото С., Фунакоши К. и др.Антитела против GM1 вызывают опосредованное комплементом разрушение кластеров натриевых каналов в периферических двигательных нервных волокнах. Дж. Neurosci . 2007 г. 11 апреля. 27 (15): 3956-67. [Медлайн].
Юки Н., Ватанабе Х, Накадзима Т., Спет П.Дж. IVIG блокирует отложение комплемента, опосредованное антителами против GM1, при мультифокальной моторной невропатии. J Neurol Neurosurg Psychiatry . 28 июля 2010 г. [Medline].
Taylor BV, Dyck PJ, Engelstad J, Gruener G, Grant I, Dyck PJ.Мультифокальная моторная нейропатия: патологические изменения в месте блокады проводимости. J Neuropathol Exp Neurol . 2004 Февраль 63 (2): 129-37. [Медлайн].
Löscher WN, Oberreiter EM, Erdler M, Quasthoff S, Culea V, Berek K и др. Мультифокальная моторная нейропатия в Австрии: общенациональное исследование клинических особенностей и реакции на лечение. Дж. Нейрол . 2018 26 сентября. [Medline].
Мияширо А., Мацуи Н., Шиматани Ю., Нодера Х., Японская группа по изучению мультифокальной моторной невропатии.Пациенты с мультифокальной моторной нейропатией недооцениваются? Эпидемиологическое обследование в Японии. Мышечный нерв . 2014 Март 49 (3): 357-61. [Медлайн].
Erdmann PG, Lindeman E, Cats EA, van den Berg LH. Функционирование пациентов с мультифокальной моторной нейропатией. J Peripher Nerv Syst . 2010 июн.15 (2): 113-9. [Медлайн].
Lange DJ, Weimer LH, Trojaborg W, et al. Мультифокальная моторная нейропатия с блокадой проводимости: медленная, но не доброкачественная. Arch Neurol . 2006 декабрь 63 (12): 1778-81. [Медлайн].
Исигаки Х., Хираиде Т., Мияги Ю., Хаяси Т., Мацубаяси Т., Симода А. и др. Мультифокальная моторная нейропатия с началом в детстве с антителами иммуноглобулина M к ганглиозидам GM1 и GM2: отчет о болезни и обзор литературы. Педиатр Нейрол . 2016 Сентябрь 62: 51-7. [Медлайн].
Bromberg MB, Franssen H. Практические правила электродиагностики при подозрении на мультифокальную моторную невропатию. J Clin Neuromuscul Dis . 2015 Март 16 (3): 141-52. [Медлайн].
Boonyapisit K, Katirji B. Мультифокальная моторная невропатия, проявляющаяся дыхательной недостаточностью. Мышечный нерв . 2000 Декабрь 23 (12): 1887-90. [Медлайн].
Straver DC, van Asseldonk JT, Notermans NC, Wokke JH, van den Berg LH, Franssen H. Холодовой парез при мультифокальной моторной невропатии. Дж. Нейрол . 2011 Февраль 258 (2): 212-7. [Медлайн]. [Полный текст].
Nobile-Orazio E, Giannotta C, Musset L, Messina P, Léger JM. Чувствительность и прогностическая ценность антител IgM к GM1 / галактоцереброзиду при мультифокальной моторной нейропатии. J Neurol Neurosurg Psychiatry . 1 августа 2013 г. [Medline].
Notturno F, Di Febo T., Yuki N, Fernandez Rodriguez BM, Corti D, Nobile-Orazio E, et al. Аутоантитела к нейрофасцину-186 и глиомедину при мультифокальной моторной нейропатии. Дж. Нейроиммунол .2014 15 ноября. 276 (1-2): 207-12. [Медлайн].
Pestronk A, Chuquilin M, Choksi R. Двигательные невропатии и связывание сывороточного IgM с дисахаридом гепарина NS6S или ганглиозидом GM1. J Neurol Neurosurg Psychiatry . 2010 июл.81 (7): 726-30. [Медлайн].
Бикман Р., ван ден Берг Л. Х., Франссен Х., Виссер Л. Х., ван Асселдонк Дж. Т., Вокке Дж. Х. Ультрасонография показывает обширное увеличение нервов при мультифокальной моторной невропатии. Неврология .2005. 65: 305-7. [Медлайн].
Vucic S, Black KR, Chong PS, Cros D. Мультифокальная моторная нейропатия: уменьшение блоков проводимости и реиннервация с длительным внутривенным иммуноглобулином. Неврология . 2004, 12 октября. 63 (7): 1264-9. [Медлайн].
Van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Мультифокальная моторная нейропатия: долгосрочная клиническая и электрофизиологическая оценка поддерживающего лечения внутривенным иммуноглобулином. Мозг .2002 августа 125 (Pt 8): 1875-86. [Медлайн].
Cats EA, van der Pol WL, Piepers S, Franssen H, Jacobs BC, van den Berg-Vos RM и др. Корреляты исхода и ответа на ВВИГ у 88 пациентов с мультифокальной моторной нейропатией. Неврология . 2010 31 августа. 75 (9): 818-25. [Медлайн].
Markvardsen LH, Harbo T. Лечение подкожным иммуноглобулином при CIDP и MMN. Эффективность, удовлетворенность лечением и стоимость. J Neurol Sci .2017 г. 15 июля. 378: 19-25. [Медлайн].
Katzberg HD, Rasutis V, Bril V. Подкожный иммуноглобулин для лечения мультифокальной моторной невропатии. Мышечный нерв . 2016 г., 54 (5): 856-863. [Медлайн].
Ruegg SJ, Fuhr P, Steck AJ. Ритуксимаб стабилизирует мультифокальную моторную невропатию, которая становится все менее чувствительной к ИГВВ. Неврология . 14 декабря 2004 г. 63 (11): 2178-9. [Медлайн].
Пестронк А., Флоренс Дж., Миллер Т. и др.Лечение полинейропатий, связанных с антителами IgM, с помощью ритуксимаба. J Neurol Neurosurg Psychiatry . 2003 апр. 74 (4): 485-9. [Медлайн].
Stieglbauer K, Topakian R, Hinterberger G, Aichner FT. Благоприятный эффект монотерапии ритуксимабом при мультифокальной моторной нейропатии. Нервно-мышечное расстройство . 2009 июл.19 (7): 473-5. [Медлайн].
[Директива] Hughes RA. Европейская федерация неврологических обществ / Руководство Общества периферических нервов по лечению мультифокальной моторной нейропатии.Отчет совместной рабочей группы Европейской федерации неврологических обществ и Общества периферических нервов. J Peripher Nerv Syst . 2006 марта 11 (1): 1-8. [Медлайн].
Nemni R, Santuccio G, Calabrese E, Galardi G, Canal N. Эффективность лечения циклоспорином при мультифокальной моторной невропатии. Дж. Нейрол . 2003 Сентябрь 250 (9): 1118-20. [Медлайн].
{Наилучшие доказательства} Умапатхи Т., Хьюз Р.А., Нобиле-Орацио Э., Леже Дж. М..Иммунодепрессанты и иммуномодуляторы для лечения мультифокальной моторной нейропатии. Кокрановская база данных Syst Rev . 2012, 18 апреля. 4: CD003217. [Медлайн].
Vlam L, Cats EA, Willemse E, Franssen H, Medic J, Piepers S, et al. Фармакокинетика внутривенного иммуноглобулина при мультифокальной моторной нейропатии. J Neurol Neurosurg Psychiatry . 2013 г. 11 декабря [Medline].
Харбо Т., Андерсен Х., Якобсен Дж. Долгосрочная терапия высокими дозами подкожного иммуноглобулина при мультифокальной моторной невропатии. Неврология . 12 октября 2010 г. 75 (15): 1377-80. [Медлайн].
Аксельсон Х.В., Оберг Дж., Аскмарк Х. Нет пользы от лечения циклофосфамидом и трансплантации аутологичных стволовых клеток крови при мультифокальной моторной невропатии. Acta Neurol Scand . 2008 июн.117 (6): 432-4. [Медлайн].
Харбо Т., Андерсен Х., Якобсен Дж. Долгосрочная терапия высокими дозами подкожного иммуноглобулина при мультифокальной моторной невропатии. Неврология .12 октября 2010 г. 75 (15): 1377-80. [Медлайн].
Lambrecq V, Krim E, Rouanet-Larrivière M, Lagueny A. Потеря чувствительности при мультифокальной моторной невропатии: клиническое и электрофизиологическое исследование. Мышечный нерв . 2009 Февраль 39 (2): 131-6. [Медлайн].
Nobile-Orazio E, Cappellari A, Priori A. Мультифокальная моторная невропатия: современные концепции и противоречия. Мышечный нерв . 31 июня 2005 г. (6): 663-80. [Медлайн].
Vlam L, van der Pol WL, Cats EA, Straver DC, Piepers S, Franssen H, et al.Мультифокальная моторная нейропатия: диагностика, патогенез и стратегии лечения. Нат Рев Нейрол . 2011 22 ноября. 8 (1): 48-58. [Медлайн].
Jongbloed BA, Haakma W, Goedee HS, Bos JW, Bos C, Hendrikse J, et al. Сравнительное исследование МРТ периферических нервов и ультразвука при мультифокальной моторной нейропатии и боковом амиотрофическом склерозе. Мышечный нерв . 2016 г., 54 (6): 1133-1135. [Медлайн].
Мультифокальная моторная нейропатия: диагностика, патогенез и стратегии лечения
Cats, E.А. и др. . Корреляты исхода и ответа на ВВИГ у 88 пациентов с мультифокальной моторной нейропатией. Неврология 75 , 818–825 (2010).
CAS PubMed PubMed Central Статья Google ученый
Чад, Д. А., Хаммер, К. и Сарджент, Дж. Медленное разрешение мультифокальной слабости и фасцикуляции: синдром обратимого двигательного нейрона. Неврология 36 , 1260–1263 (1986).
CAS Статья Google ученый
Рот, Г., Рор, Дж., Магистрис, М. Р. и Окснер, Ф. Двигательная невропатия с проксимальной многоочаговой стойкой блокадой проводимости, фасцикуляциями и миокимией. Эволюция до тетраплегии. Eur. Neurol. 25 , 416–423 (1986).
CAS Статья Google ученый
Парри, Дж. Дж. И Кларк, С.Мультифокальная приобретенная демиелинизирующая нейропатия, маскирующаяся под заболевание двигательных нейронов. Мышечный нерв 11 , 103–107 (1988).
CAS Статья Google ученый
Pestronk, A. et al. . Мультифокальная моторная невропатия, поддающаяся лечению с помощью антител к ганглиозиду GM1. Ann. Neurol. 24 , 73–78 (1988).
CAS Статья Google ученый
Пестронк, А. и др. . Синдромы нижних мотонейронов, определяемые паттернами слабости, нарушениями нервной проводимости и высокими титрами антигликолипидных антител. Ann. Neurol. 27 , 316–326 (1990).
CAS Статья Google ученый
Ланге, Д. Дж. и др. . Мультифокальная моторная нейропатия с блокадой проводимости: это отдельная клиническая форма? Неврология 42 , 497–505 (1992).
CAS Статья Google ученый
Bouche, P. и др. . Мультифокальная моторная нейропатия с блокадой проводимости: исследование 24 пациентов. J. Neurol. Нейрохирург. Психиатрия. 59 , 38–44 (1995).
CAS PubMed PubMed Central Статья Google ученый
Джасперт, А., Клаус, Д., Грел, Х. и Нойндорфер, Б.Мультифокальная моторная нейропатия: клинические и электрофизиологические данные. J. Neurol. 243 , 684–692 (1996).
CAS Статья Google ученый
Ле Форестье, Н. и др. . Мультифокальные моторные невропатии с блокадой проводимости. 39 кейсов. Rev. Neurol. (Париж) 153 , 579–586 (1997).
CAS Google ученый
Бентес, К., де Карвальо, М., Евангелиста, Т. и Сэйлс-Луис, М. Л. Мультифокальная моторная невропатия, имитирующая заболевание двигательных нейронов: девять случаев. J. Neurol. Sci. 169 , 76–79 (1999).
CAS Статья Google ученый
Тейлор, Б.В., Райт, Р.А., Харпер, К.М. и Дайк, П.Дж. Естественный анамнез 46 пациентов с мультифокальной моторной нейропатией с блокадой проводимости. Мышечный нерв 23 , 900–908 (2000).
CAS Статья Google ученый
Азулай, Дж. П. и др. . Внутривенное введение иммуноглобулинов у пациентов с синдромами двигательных нейронов, связанными с антителами против GM1: двойное слепое плацебо-контролируемое исследование. Неврология 44 , 429–432 (1994).
CAS Статья Google ученый
Ван ден Берг, Л.Х. и др. . Лечение мультифокальной моторной нейропатии высокими дозами внутривенных иммуноглобулинов: двойное слепое плацебо-контролируемое исследование. J. Neurol. Нейрохирург. Психиатрия 59 , 248–252 (1995).
CAS PubMed PubMed Central Статья Google ученый
Federico, P., Zochodne, D. W., Hahn, A. F., Brown, W. F. и Feasby, T. E. Мультифокальная моторная невропатия, улучшенная с помощью IVIg: рандомизированное, двойное слепое, плацебо-контролируемое исследование. Неврология 55 , 1256–1262 (2000).
CAS Статья Google ученый
Леже, Дж. М. и др. . Внутривенная иммуноглобулиновая терапия при мультифокальной моторной нейропатии: двойное слепое плацебо-контролируемое исследование. Мозг 124 , 145–153 (2001).
CAS Статья Google ученый
Ван ден Берг-Вос, Р.М., Франссен, Х., Вокке, Дж. Х. и Ван ден Берг, Л. Х. Мультифокальная моторная невропатия: долгосрочная клиническая и электрофизиологическая оценка поддерживающего лечения внутривенным иммуноглобулином. Мозг 125 , 1875–1886 (2002).
CAS Статья Google ученый
Теренги, Ф. и др. . Как долго внутривенные иммуноглобулины эффективны при мультифокальной моторной нейропатии? Неврология 62 , 666–668 (2004).
CAS Статья Google ученый
Нобиле-Орацио, Э. Мультифокальная моторная нейропатия. J. Neuroimmunol. 115 , 4–18 (2001).
CAS Статья Google ученый
Сли М., Селван А. и Донахи М. Мультифокальная моторная нейропатия: спектр диагностики и ответ на лечение. Неврология 69 , 1680–1687 (2007).
Артикул Google ученый
Ван Асселдонк, Дж. Т. и др. . Демиелинизация и потеря аксонов при мультифокальной моторной нейропатии: распределение и связь со слабостью. Мозг 126 , 186–198 (2003).
CAS Статья Google ученый
Ван ден Берг-Вос, Р. М. и др. . Тяжесть заболевания при мультифокальной моторной нейропатии и ее связь с ответом на лечение иммуноглобулином. J. Neurol. 249 , 330–336 (2002).
CAS Статья Google ученый
Straver, D. C. и др. . Холодовой парез при мультифокальной моторной нейропатии. J. Neurol. 258 , 212–217 (2011).
Артикул Google ученый
О’Лири, К. П., Манн, А. С., Лох, Дж. И Уиллисон, Х. Дж. Гипертрофия мышц при мультифокальной моторной невропатии связана с непрерывной активностью двигательных единиц. Мышечный нерв 20 , 479–485 (1997).
CAS Статья Google ученый
Delmont, E. и др. . Имеются ли пациенты, у которых наблюдается снижение амплитуды SNAP во время курса MMN, с другим заболеванием? J. Neurol. 256 , 1876–1880 (2009).
Артикул Google ученый
Ламбрек, В., Крим, Э., Руане-Ларривьер, М. и Лагуэни, А. Потеря чувствительности при мультифокальной моторной невропатии: клиническое и электрофизиологическое исследование. Мышечный нерв 39 , 131–136 (2009).
Артикул Google ученый
Ливенс, И. и др. . Мультифокальная моторная нейропатия: ретроспективное исследование скорости проводимости сенсорных нервов при длительном наблюдении за 21 пациентом. Rev. Neurol. (Париж) 165 , 243–248 (2009).
CAS Статья Google ученый
Совместная оперативная группа EFNS и PNS. Руководство Европейской федерации неврологических обществ / Общества периферических нервов по ведению мультифокальной моторной нейропатии: отчет совместной рабочей группы Европейской федерации неврологических обществ и Общества периферических нервов — первая редакция. Дж. Перифер. Nerv. Syst. 15 , 295–301 (2010).
Прингл, К.Э., Белден, Дж., Вейтч, Дж. Э. и Браун, У. Ф. Мультифокальная моторная невропатия, проявляющаяся как офтальмоплегия. Мышечный нерв 20 , 347–351 (1997).
CAS Статья Google ученый
Кадзи Р., Шибасаки Х. и Кимура Дж. Мультифокальная демиелинизирующая моторная нейропатия: поражение черепных нервов и иммуноглобулиновая терапия. Неврология 42 , 506–509 (1992).
CAS Статья Google ученый
Кириакидес Т., Papacostas, S., Papanicolaou, E., Bagdades, E. & Papathanasiou, E. S. Синдром гиповентиляции сна и дыхательная недостаточность из-за мультифокальной моторной нейропатии с блокадой проводимости. Мышечный нерв 43 , 610–614 (2011).
Артикул Google ученый
Бейдун, С. Р. и Коупленд, Д. Двусторонняя диафрагмальная невропатия как характерный признак мультифокальной моторной нейропатии с блокадой проводимости. Мышечный нерв 23 , 556–559 (2000).
CAS Статья Google ученый
Boonyapisit, K. & Katirji, B. Мультифокальная моторная невропатия, проявляющаяся дыхательной недостаточностью. Мышечный нерв 23 , 1887–1890 (2000).
CAS Статья Google ученый
Кавалетти, Г. и др. . Быстро прогрессирующая мультифокальная моторная нейропатия с параличом диафрагмального нерва: эффект ночной вспомогательной вентиляции. J. Neurol. 245 , 613–616 (1998).
CAS Статья Google ученый
Ван Асселдонк, Дж. Т. и др. . Потеря аксонов — важный фактор, определяющий слабость при мультифокальной моторной нейропатии. J. Neurol. Нейрохирург. Психиатрия 77 , 743–747 (2006).
CAS PubMed PubMed Central Статья Google ученый
ван ден Берг-Вос, Р.М. и др. . Спорадическое заболевание нижних мотонейронов с началом у взрослых: классификация подтипов. Мозг 126 , 1036–1047 (2003).
CAS Статья Google ученый
Трейнор, Б. Дж. и др. . Синдромы, имитирующие боковой амиотрофический склероз: популяционное исследование. Arch. Neurol. 57 , 109–113 (2000).
CAS PubMed PubMed Central Статья Google ученый
Донахи, М.Классификация и клинические особенности заболеваний двигательных нейронов и двигательных невропатий у взрослых. J. Neurol. 246 , 331–333 (1999).
CAS Статья Google ученый
Visser, J. et al. . Мимические синдромы в спорадических случаях прогрессирующей мышечной атрофии позвоночника. Неврология 58 , 1593–1596 (2002).
CAS Статья Google ученый
Ван Асселдонк, Дж.Т. и др. . Демиелинизация и потеря аксонов при мультифокальной моторной нейропатии: распределение и связь со слабостью. Мозг 126 , 186–198 (2003).
CAS Статья Google ученый
Ван ден Берг-Вос, Р. М., Франссен, Х., Вокке, Дж. Х., Ван Эс, Х. У. и Ван ден Берг, Л. Х. Мультифокальная моторная невропатия: диагностические критерии, которые предсказывают ответ на лечение иммуноглобулином. Ann.Neurol. 48 , 919–926 (2000).
CAS Статья Google ученый
Совместная оперативная группа EFNS и PNS. Руководство Европейской федерации неврологических обществ / Общества периферических нервов по ведению хронической воспалительной демиелинизирующей полирадикулоневропатии: отчет совместной рабочей группы Европейской федерации неврологических обществ и Общества периферических нервов — первая редакция. Дж.Периферия. Nerv. Syst. 15 , 1–9 (2010).
Льюис, Р. А., Самнер, А. Дж., Браун, М. Дж., Эсбери, А. К. Мультифокальная демиелинизирующая невропатия со стойкой блокадой проводимости. Неврология 32 , 958–964 (1982).
CAS PubMed PubMed Central Статья Google ученый
Парри, Г. Дж. Являются ли мультифокальная моторная невропатия и синдром Льюиса – Самнера отдельными нозологическими объектами? Мышечный нерв 22 , 557–559 (1999).
CAS Статья Google ученый
Ван ден Берг-Вос, Р. М. и др. . Мультифокальная воспалительная демиелинизирующая нейропатия: отдельное клиническое проявление? Неврология 54 , 26–32 (2000).
CAS Статья Google ученый
О, С. Дж., Клауссен, Г. С. и Ким, Д. С. Мультиплексная моторная и сенсорная демиелинизирующая мононевропатия (мультифокальная моторная и сенсорная демиелинизирующая нейропатия): отдельное образование или вариант хронической воспалительной демиелинизирующей полинейропатии? Дж.Периферия. Nerv. Syst. 2 , 362–369 (1997).
CAS Google ученый
Горсон, К. К., Роппер, А. Х. и Вайнберг, Д. Х. Многоочаговая хроническая воспалительная демиелинизирующая полинейропатия с преобладанием верхних конечностей. Мышечный нерв 22 , 758–765 (1999).
CAS Статья Google ученый
Льюис, Р.А. Мультифокальная моторная невропатия и синдром Льюиса Самнера: две разные сущности. Мышечный нерв 22 , 1738–1739 (1999).
CAS Статья Google ученый
Саперштейн, Д. С. и др. . Мультифокальная приобретенная демиелинизирующая сенсорная и моторная нейропатия: синдром Льюиса-Самнера. Мышечный нерв 22 , 560–566 (1999).
CAS PubMed PubMed Central Статья Google ученый
Саперштейн, Д.С., Кац, Дж. С., Амато, А. А. и Барон, Р. Дж. Клинический спектр хронических приобретенных демиелинизирующих полинейропатий. Мышечный нерв 24 , 311–324 (2001).
CAS Статья Google ученый
Виала, К. и др. . Последующее исследование и ответ на лечение у 23 пациентов с синдромом Льюиса – Самнера. Мозг 127 , 2010–2017 (2004).
CAS Статья Google ученый
Verschueren, A. и др. . Синдром Льюиса – Самнера и мультифокальная моторная нейропатия. Мышечный нерв 31 , 88–94 (2005).
Артикул Google ученый
Льюис Р.А. Невропатии, связанные с блокадой проводимости. Curr. Opin. Neurol. 20 , 525–530 (2007).
Артикул Google ученый
Нобиле-Орацио, Э., Каппеллари, А.& Priori, A. Мультифокальная моторная невропатия: современные концепции и противоречия. Мышечный нерв 31 , 663–680 (2005).
Артикул Google ученый
Beydoun, S.R. Мультифокальная моторная невропатия с блокадой проводимости, ошибочно диагностированной как множественная невропатия с захватом. Мышечный нерв 21 , 813–815 (1998).
CAS Статья Google ученый
Хьюз, П.R. 79 th Международный семинар ENMC: мультифокальная моторная невропатия. 14–15 апреля 2000 г., Хилверсюм, Нидерланды. Neuromuscul. Disord. 11 , 309–314 (2001).
CAS Статья Google ученый
Олни, Р. К., Льюис, Р. А., Патнэм, Т. Д., Кампеллон, Дж. В. Младший и Американская ассоциация электродиагностической медицины. Критерии согласия для диагностики мультифокальной моторной нейропатии. Мышечный нерв 27 , 117–121 (2003).
Артикул Google ученый
ван Шайк, И. Н. и др. . Руководство Европейской федерации неврологических обществ / Общества периферических нервов по лечению мультифокальной моторной нейропатии. Eur. J. Neurol. 13 , 802–808 (2006).
CAS Статья Google ученый
Ван Асселдонк, Дж.Т., Ван ден Берг, Л. Х., Винеке, Г. Х., Вокке, Дж. Х. и Франссен, Х. Критерии блокады проводимости на основе компьютерных симуляционных исследований нервной проводимости с данными человека, полученными в сегменте срединного нерва предплечья. Мозг 129 , 2447–2460 (2006).
CAS Статья Google ученый
Ван Асселдонк, Дж. Т., Франссен, Х., Ван ден Берг-Вос, Р. М., Вокке, Дж. Х. и Ван ден Берг, Л.H. Мультифокальная моторная невропатия. Lancet Neurol. 4 , 309–319 (2005).
Артикул Google ученый
Delmont, E. и др. . Мультифокальная моторная нейропатия с блокадой проводимости и без нее: единое целое? Неврология 67 , 592–596 (2006).
CAS Статья Google ученый
Чаудри, В.& Swash, M. Мультифокальная моторная нейропатия: необходима ли блокада проводимости? Неврология 67 , 558–559 (2006).
Артикул Google ученый
Menkes, D. L. Мультифокальная моторная нейропатия с блокадой проводимости и без нее: единое целое? Неврология 68 , 1161–1162 (2007).
Артикул Google ученый
Менкес, Д.Л., Худ, Д. К., Баллестерос, Р. А. и Уильямс, Д. А. Стимуляция корней улучшает выявление приобретенных демиелинизирующих полинейропатий. Мышечный нерв 21 , 298–308 (1998).
CAS Статья Google ученый
Arunachalam, R., Osei-Lah, A. & Mills, K. R. Чрескожная стимуляция корня шейки матки в диагностике мультифокальной моторной нейропатии с блокадой проводимости. J. Neurol. Нейрохирург.Психиатрия. 74 , 1329–1331 (2003).
CAS PubMed PubMed Central Статья Google ученый
Attarian, S., Azulay, J. P., Verschueren, A. & Pouget, J. Магнитная стимуляция с использованием техники тройной стимуляции у пациентов с мультифокальной нейропатией без блокады проводимости. Мышечный нерв 32 , 710–714 (2005).
Артикул Google ученый
Деруид, Н. и др. . Техника тройной стимуляции при мультифокальной нейропатии с блокадой проводимости. Мышечный нерв 35 , 632–636 (2007).
Артикул Google ученый
Каджи, Р. и др. . Активно-зависимая блокада проведения при мультифокальной моторной нейропатии. Мозг 123 , 1602–1611 (2000).
Артикул Google ученый
Nodera, H. и др. . Активно-зависимый блок проводимости при мультифокальной моторной нейропатии: тест на магнитную усталость. Неврология 67 , 280–287 (2006).
CAS Статья Google ученый
Hitomi, T. и др. . Динамическое изменение проксимальной проводимости при демиелинизирующих невропатиях: цервикальная магнитная стимуляция в сочетании с максимальным произвольным сокращением. Clin. Neurophysiol. 118 , 741–750 (2007).
Артикул Google ученый
Стравер, Д. К., ван ден Берг, Л. Х., ван ден Берг-Вос, Р. М. и Франссен, Х. Активно-зависимый блок проводимости при мультифокальной моторной невропатии. Мышечный нерв 43 , 31–36 (2011).
Артикул Google ученый
Parry, G. J. Отчет о клиническом случае AAEM № 30: мультифокальная моторная невропатия. Мышечный нерв 19 , 269–276 (1996).
CAS Статья Google ученый
van Schaik, I. N., Bossuyt, P. M., Brand, A. & Vermeulen, M. Диагностическая ценность антител GM1 при нарушениях двигательных нейронов и невропатиях: метаанализ. Неврология 45 , 1570–1577 (1995).
CAS Статья Google ученый
Тейлор, Б.В., Гросс, Л. и Виндебанк, А.J. Чувствительность и специфичность тестирования антител против GM1. Неврология 47 , 951–955 (1996).
CAS Статья Google ученый
Кошки, Э. А. и др. . Мультифокальная моторная нейропатия: ассоциация антител IgM к GM1 с клиническими особенностями. Неврология 75 , 1961–1967 (2010).
CAS Статья Google ученый
Уиллисон, Х.Дж. И Юки Н. Периферические невропатии и антигликолипидные антитела. Мозг 125 , 2591–2625 (2002).
Артикул Google ученый
Нобиле-Орацио, Э. и др. . Насколько полезны антинейральные антитела IgM в диагностике хронических иммуноопосредованных невропатий? J. Neurol. Sci. 266 , 156–163 (2008).
CAS Статья Google ученый
Гуч, К.L. & Amato, A. A. Имеют ли анти-ганглиозидные антитела клиническую ценность при мультифокальной моторной нейропатии? Неврология 75 , 1950–1951 (2010).
Артикул Google ученый
Ван Эс, Х. В. и др. . Магнитно-резонансная томография плечевого сплетения у пациентов с мультифокальной моторной нейропатией. Неврология 48 , 1218–1224 (1997).
CAS Статья Google ученый
Эчаниз-Лагуна, А.И Дитеманн, Дж. Л. Видя блоки: МРТ плечевого сплетения при мультифокальной моторной невропатии. J. Neurol. Нейрохирург. Психиатрия 82 , 728 (2011).
Артикул Google ученый
Бикман, Р. и др. . Ультрасонография показывает обширное увеличение нервов при мультифокальной моторной невропатии. Неврология 65 , 305–307 (2005).
CAS Статья Google ученый
Приори, А. и др. . Характерные аномалии прочностных свойств моторных аксонов при мультифокальной моторной нейропатии и болезни моторных нейронов. Мозг 125 , 2481–2490 (2002).
CAS Статья Google ученый
Кирнан, М. К., Гульельми, Дж. М., Каджи, Р., Мюррей, Н. М. и Босток, Х. Доказательства гиперполяризации аксональной мембраны при мультифокальной моторной нейропатии с блокадой проводимости. Мозг 125 , 664–675 (2002).
Артикул Google ученый
Каджи Р. Физиология блока проводимости при мультифокальной моторной невропатии и других демиелинизирующих невропатиях. Мышечный нерв 27 , 285–296 (2003).
Артикул Google ученый
Priori, A. и др. . Патофизиологическая неоднородность блоков проведения при мультифокальной моторной нейропатии. Мозг 128 , 1642–1648 (2005).
Артикул Google ученый
Ауэр, Р. Н., Белл, Р. Б. и Ли, М. А. Невропатия с образованием луковиц и чисто моторными проявлениями. банка. J. Neurol. Sci. 16 , 194–197 (1989).
CAS Статья Google ученый
Каджи, Р. и др. . Патологические находки в месте блокады проводимости при мультифокальной моторной нейропатии. Ann. Neurol. 33 , 152–158 (1993).
CAS Статья Google ученый
Corbo, M. и др. . Исследования биопсии двигательного нерва при двигательной невропатии и заболевании двигательных нейронов. Мышечный нерв 20 , 15–21 (1997).
CAS Статья Google ученый
Тейлор Б.В. и др. . Мультифокальная моторная нейропатия: патологические изменения в месте блокады проводимости. J. Neuropathol. Exp. Neurol. 63 , 129–137 (2004).
Артикул Google ученый
Вучич, С., Блэк, К., Чонг, П. С. и Крос, Д. Мультифокальная моторная невропатия с блокадой проведения: распространение демиелинизации и дегенерации аксонов. Clin. Neurophysiol. 118 , 124–130 (2007).
Артикул Google ученый
Вучич, С., Блэк, К. Р., Чонг, П. С. и Крос, Д. Мультифокальная моторная нейропатия: снижение проводимости и реиннервация с длительным введением иммуноглобулина IV. Неврология 63 , 1264–1269 (2004).
Артикул Google ученый
Огава-Гото, К., Фунамото, Н., Охта, Ю., Абэ, Т. и Нагашима, К. Миелиновые ганглиозиды периферической нервной системы человека: обогащение GM1 миелином двигательного нерва, выделенным из конский хвост. J. Neurochem. 59 , 1844–1849 (1992).
CAS Статья Google ученый
Susuki, K. и др. . Ганглиозиды способствуют стабильности паранодальных соединений и кластеров ионных каналов в миелинизированных нервных волокнах. Glia 55 , 746–757 (2007).
Артикул Google ученый
Сусуки, К. и др. . Антитела против GM1 вызывают опосредованное комплементом разрушение кластеров натриевых каналов в периферических двигательных нервных волокнах. J. Neurosci. 27 , 3956–3967 (2007).
CAS Статья Google ученый
Арасаки К., Кусуноки С., Кудо Н. и Канадзава И. Острая блокада проводимости in vitro после воздействия антиганглиозидной сыворотки. Мышечный нерв 16 , 587–593 (1993).
CAS Статья Google ученый
Uncini, A., Santoro, M., Corbo, M., Lugaresi, A. & Latov, N. Нарушения проводимости, вызванные сыворотками пациентов с мультифокальной моторной невропатией и антителами против GM1. Мышечный нерв 16 , 610–615 (1993).
CAS Статья Google ученый
Робертс, М., Уиллисон, Х. Дж., Винсент, А. и Ньюсом-Дэвис, Дж. Мультифокальная моторная нейропатия Сыворотка человека блокирует проведение дистальных моторных нервов у мышей. Ann. Neurol. 38 , 111–118 (1995).
CAS Статья Google ученый
Харви, Г. К. и др. . Неспособность анти-GM1 IgG или IgM вызвать блокаду проводимости после внутринейрального переноса. Мышечный нерв 18 , 388–394 (1995).
CAS Статья Google ученый
Хирота, Н. и др. . Физиологический эффект анти-GM1 антител на скачкообразную проводимость и трансмембранные токи в одиночных моторных аксонах. Мозг 120 , 2159–2169 (1997).
Артикул Google ученый
Юки, Н., Ватанабе, Х., Накадзима, Т. и Спат, П. Дж. IVIG блокирует отложение комплемента, опосредованное антителами против GM1, при мультифокальной моторной невропатии. J. Neurol. Нейрохирург. Психиатрия 82 , 87–91 (2011).
CAS Статья Google ученый
Пиперс, С. и др. . IVIg ингибирует активность классического пути и опосредованное IgM отложение комплемента в MMN. J. Neuroimmunol. 229 , 256–262 (2010).
CAS Статья Google ученый
Джейкобс, Б. С. и др. . Иммуноглобулины подавляют патофизиологические эффекты анти-GQ1b-положительной сыворотки на окончаниях двигательного нерва путем ингибирования связывания антител. Мозг 126 , 2220–2234 (2003).
Артикул Google ученый
Холстед, С. К. и др. . Экулизумаб предотвращает нейропатию, опосредованную антиганглиозидными антителами, на мышиной модели. Мозг 131 , 1197–1208 (2008).
Артикул Google ученый
Джейкобс, Б. С. и др. . Campylobacter jejuni инфекции и антитела против GM1 при синдроме Гийена-Барре. Ann. Neurol. 40 , 181–187 (1996).
CAS PubMed PubMed Central Статья Google ученый
Теренги, Ф., Аллария, С., Скарлато, Г. и Нобиле-Орацио, Е. Мультифокальная моторная невропатия и Campylobacter jejuni реактивность. Неврология 59 , 282–284 (2002).
Артикул Google ученый
Эурелингс, М. и др. . Антиганглиозидные антитела при полинейропатии, связанной с моноклональной гаммопатией. Неврология 57 , 1909–1912 (2001).
CAS Статья Google ученый
Кайда К. и др. . Комплексы ганглиозидов как новые целевые антигены при синдроме Гийена – Барре. Ann. Neurol. 56 , 567–571 (2004).
CAS Статья Google ученый
Нобиле-Орацио, Э., Джаннотта, С. и Бриани, С. Антитела IgM к ганглиозидному комплексу при мультифокальной моторной невропатии и хронических иммуноопосредованных невропатиях. J. Neuroimmunol. 219 , 119–122 (2010).
CAS Статья Google ученый
Pestronk, A., Choksi, R., Blume, G. & Lopate, G. Мультифокальная моторная невропатия: связывание сывороточного IgM с липидной смесью, содержащей ганглиозиды GM1, но не только с GM1. Неврология 48 , 1104–1106 (1997).
CAS Статья Google ученый
Пестронк, А., Чукилин, М. и Чокси, Р. Двигательные невропатии и связывание сывороточного IgM с дисахаридом гепарина NS6S или ганглиозидом GM1. J. Neurol. Нейрохирург. Психиатрия. 81 , 726–730 (2010).
Артикул Google ученый
van der Pol, W. L., Cats, E. A. и van den Berg, L.H. Внутривенное введение иммуноглобулинов при мультифокальной моторной нейропатии. J. Clin. Иммунол. 30 (Приложение 1), S79 – S83 (2010).
CAS Статья Google ученый
Кляйн, Дж. И Сато, А. Система HLA: первая из двух частей. N. Engl. J. Med. 343 , 702–709 (2000).
CAS PubMed PubMed Central Статья Google ученый
Сутеджа, Н.А. и др. . Повышенная частота HLA-DRB1 * 15 у пациентов с мультифокальной моторной нейропатией. Неврология 74 , 828–832 (2010).
CAS Статья Google ученый
Шмидт, Х., Уильямсон, Д. и Эшли-Кох, А. HLA-DR15 гаплотип и рассеянный склероз: обзор HuGE. Am. J. Epidemiol. 165 , 1097–1109 (2007).
Артикул Google ученый
Влам, Л. и др. . Мультифокальная моторная нейропатия не связана с генетической изменчивостью в генах PTPN22, BANK1, Blk, FCGR2B, CD1A / E и TAG-1 . Дж. Перифер. Nerv. Syst. 16 , 175–179 (2011).
CAS Статья Google ученый
van Schaik, I. N., van den Berg, L.H., de Haan, R. & Vermeulen, M. Внутривенный иммуноглобулин для лечения мультифокальной моторной невропатии. Кокрановская база данных систематических обзоров , выпуск 2.Изобразительное искусство. №: CD004429. http://dx.doi.org/10.1002/14651858.CD004429.pub2 (2005).
Леже, Дж. М. и др. . Внутривенный иммуноглобулин как краткосрочная и долгосрочная терапия мультифокальной моторной нейропатии: ретроспективное исследование ответа на IVIg и его прогностических критериев у 40 пациентов. J. Neurol. Нейрохирург. Психиатрия 79 , 93–96 (2008).
Артикул Google ученый
Бауманн, А., Hess, C. W. & Sturzenegger, M. Увеличение дозы IVIg при мультифокальной моторной нейропатии: проспективное шестимесячное наблюдение. J. Neurol. 256 , 608–614 (2009).
CAS Статья Google ученый
Меркис, И. С. и Шмитц, П. И. Приближение к пациентам: общая сумма баллов по инвалидности по INCAT лучше соотносится с собственными клиническими суждениями пациентов об иммуно-опосредованных полинейропатиях. Дж.Neurol. Нейрохирург. Психиатрия 77 , 970–972 (2006).
CAS PubMed PubMed Central Статья Google ученый
Leger, J. M. & Salachas, F. Диагностика моторной невропатии. Eur. J. Neurol. 8 , 201–208 (2001).
CAS Статья Google ученый
Кошки, Э. А., ван дер Поль, В. Л., Бертенс, А.S. & van den Berg, L.H. Лечение внутривенных иммуноглобулинов в домашних условиях удобно и экономит время у пациентов с мультифокальной моторной нейропатией. Дж. Перифер. Nerv. Syst. 16 , 147–149 (2011).
Артикул Google ученый
Ефтимов, Ф., Вермёлен, М., де Хаан, Р. Дж., Ван ден Берг, Л. Х. и ван Шайк, И. Н. Подкожная иммуноглобулиновая терапия при мультифокальной моторной невропатии. Дж. Перифер. Nerv. Syst. 14 , 93–100 (2009).
CAS Статья Google ученый
Харбо, Т. и др. . Подкожный иммуноглобулин по сравнению с внутривенным при мультифокальной моторной нейропатии: рандомизированное одно-слепое перекрестное исследование. Eur. J. Neurol. 16 , 631–638 (2009).
CAS Статья Google ученый
Мисбах, С.А. и др. . Протокол плавного перехода для пациентов с мультифокальной моторной нейропатией от внутривенной к подкожной иммуноглобулиновой терапии: открытое исследование, подтверждающее правильность концепции. Дж. Перифер. Nerv. Syst. 16 , 92–97 (2011).
CAS Статья Google ученый
Харбо Т., Андерсен Х. и Якобсен Дж. Длительная терапия высокими дозами подкожного иммуноглобулина при мультифокальной моторной нейропатии. Неврология 75 , 1377–1380 (2010).
CAS Статья Google ученый
Ван ден Берг, Л. Х., Франссен, Х. и Вокке, Дж. Х. Долгосрочный эффект лечения внутривенным иммуноглобулином при мультифокальной моторной нейропатии. Мозг 121 , 421–428 (1998).
Артикул Google ученый
Ван ден Берг, Л.Х., Локхорст, Х. и Вокке, Дж. Х. Импульсные высокие дозы дексаметазона не эффективны у пациентов с мультифокальной моторной нейропатией. Неврология 48 , 1135 (1997).
CAS Статья Google ученый
Карпо, М. и др. . Ухудшение мультифокальной моторной нейропатии после плазмафереза. Неврология 50 , 1480–1482 (1998).
CAS Статья Google ученый
Леманн, Х.С. и др. . Клиническое значение терапевтического плазмафереза при мультифокальной моторной нейропатии. J. Neurol. Sci. 271 , 34–39 (2008).
CAS Статья Google ученый
Ефтимов Ф. и Ван Шайк И. Н. Иммунотерапия мультифокальной моторной нейропатии. Мнение эксперта. Биол. Ther. 11 , 329–342 (2011).
CAS Статья Google ученый
Умапатхи, Т., Hughes, R.A., Nobile-Orazio, E. & Leger, J.M. Иммунодепрессанты и иммуномодуляторы для лечения мультифокальной моторной нейропатии. Кокрановская база данных систематических обзоров , выпуск 1, ст. №: CD003217. http://dx.doi.org/10.1002/14651858.CD003217.pub3 (2009 г.).
Нобиле-Орацио, Э., Галлия, Ф., Туччилло, Ф. и Теренги, Ф. Хроническая воспалительная демиелинизирующая полирадикулоневропатия и мультифокальная моторная нейропатия: обновление лечения. Curr. Opin. Neurol. 23 , 519–523 (2010).
CAS Статья Google ученый
Пиперс, С. и др. . Микофенолятмофетил в качестве дополнительной терапии для пациентов с MMN: рандомизированное контролируемое исследование. Мозг 130 , 2004–2010 (2007).
Артикул Google ученый
Фитцпатрик А. М. и др. . Открытое клиническое испытание ингибирования комплемента при мультифокальной моторной невропатии. Дж. Перифер. Nerv. Syst. 16 , 84–91 (2011).
PubMed PubMed Central Статья Google ученый
Pestronk, A. et al. . Лечение полинейропатий, связанных с антителами IgM, с помощью ритуксимаба. J. Neurol. Нейрохирург. Психиатрия 74 , 485–489 (2003).
CAS PubMed PubMed Central Статья Google ученый
Рохас-Гарсия, Р. и др. . Хроническая невропатия с анти-ганглиозидными антителами IgM: отсутствие длительного ответа на ритуксимаб. Неврология 61 , 1814–1816 (2003).
CAS Статья Google ученый
Ruegg, S.J., Fuhr, P. & Steck, A.J. Ритуксимаб стабилизирует мультифокальную моторную невропатию, которая становится все менее чувствительной к IVIg. Неврология 63 , 2178–2179 (2004).
Артикул Google ученый
Горсон, К.