Патоморфологическая диагностика эрозивно-язвенного рефлюкс-эзофагита по биопсии слизистой оболочки пищевода
Актуальность проблемы гастроэзофагеальной рефлюксной болезни (ГЭРБ) обусловлена ее высокой распространенностью, которая, по данным ряда авторов, среди взрослого населения достигает 20—40% [1]. Особое место среди форм ГЭРБ занимают эрозивно-язвенные поражения, которые могут осложняться перфорацией, рубцовой стриктурой пищевода, приводить к медиастиниту и являться причиной смерти. Для их характеристики обычно используют эндоскопические классификации, учитывающие распространенность поражений к общей площади дистального отдела пищевода. Сведения о гистологической характеристике и морфогенезе острых язв и эрозий слизистой оболочки пищевода (СОП) остаются неполными [2, 3], в то время как аналогичные изменения в желудке и двенадцатиперстной кишке хорошо изучены и подробно описаны [4—6]. В то же время знание особенностей развития и заживления деструктивных поражений СОП под влиянием содержимого желудка и/или двенадцатиперстной кишки должно способствовать разработке особых методов лечения, с использованием, помимо антисекреторных, противовоспалительных препаратов и цитопротекторов слизистой оболочки, синергистов или антагонистов факторов роста.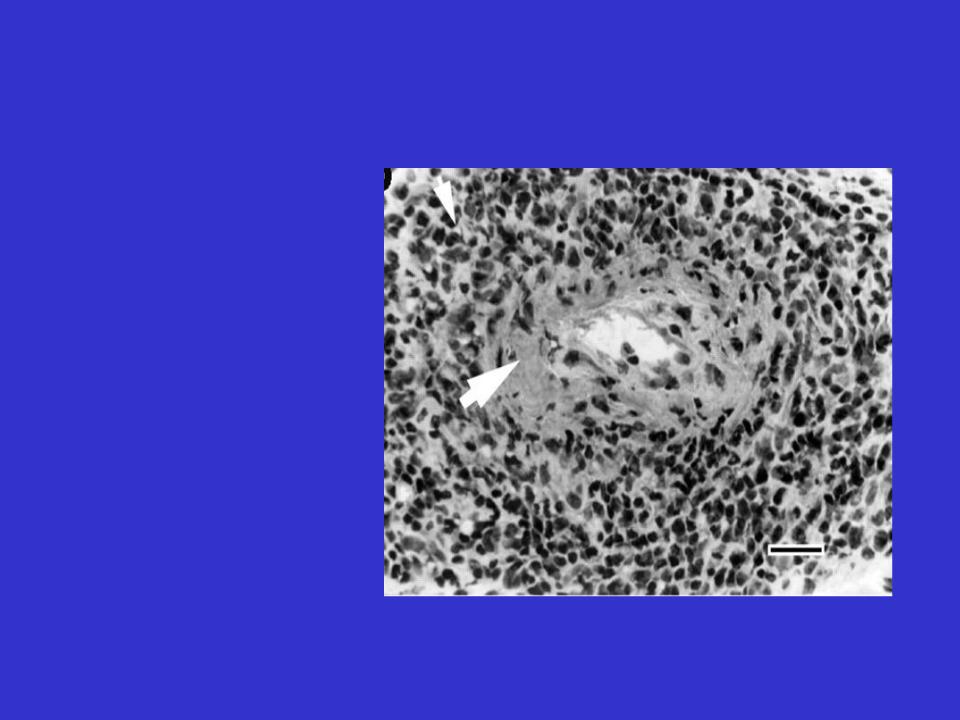
Цель исследования — изучить морфологическую характеристику и морфогенез эрозивно-язвенных поражений при ГЭРБ.
В работе использовали материал 2220 биоптатов слизистой оболочки дистального отдела пищевода и области сквамозно-цилиндроклеточного соединения (СЦС) у 669 пациентов (у 90 больных проводились повторные исследования в динамике лечения ИПП). Гистологические препараты окрашивали гематоксилином и эозином, по Ван-Гизону, метиленовым синим по Лефлеру, комбинированным методом — алциановым синим и PAS-реакцией.
Пептические эрозии выявлены в 24% наблюдений, язвы — в 17%. Они, как правило, локализовались в области СЦС или зоны, выстланной многослойным плоским эпителием (МПЭ). При эрозиях дефекты ограничивались слизистой оболочкой до мышечной пластинки, для язв характерно было проникновение в подслизистый и мышечный слои. Микроскопически эрозии в биоптатах не всегда легко выявить в связи с тем, что они маскируются регенерирующим, резко утолщенным МПЭ.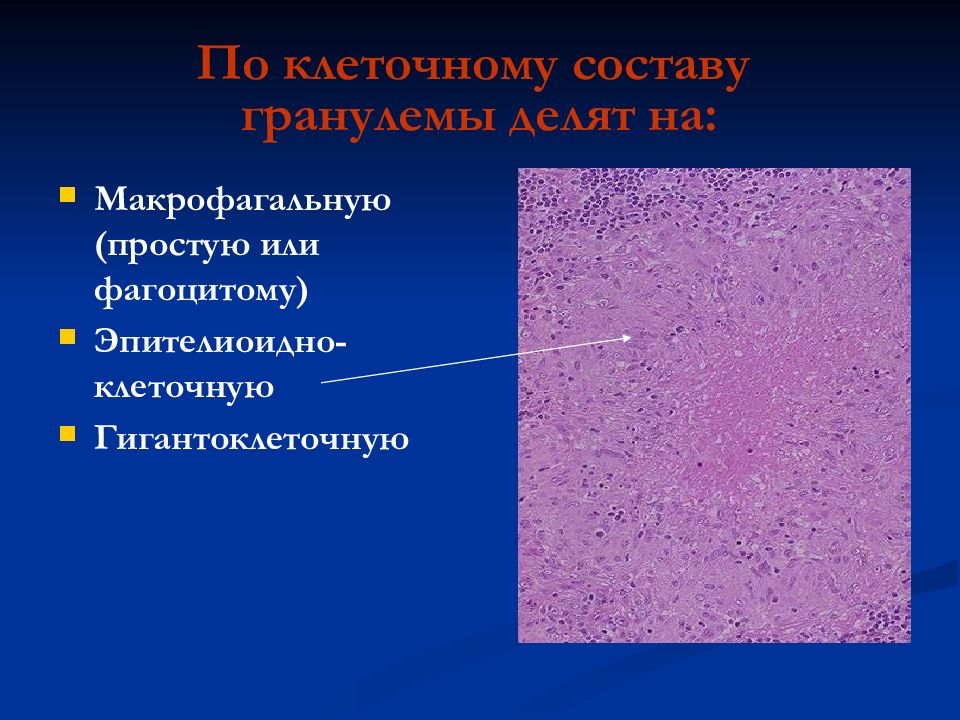
Отличительный признак острых пептических поражений — возникновение своеобразного некроза коллагеновых и мышечных волокон, который в литературе часто обозначают термином «фибриноидный» некроз. На самом деле в тех единичных случаях, когда удается увидеть начальные стадии повреждения СОП желудочным соком, типичной картины «фибриноида» не наблюдается. Покровный МПЭ обычно некротизируется и отслаивается на значительном протяжении пластами или ограниченно, с образованием узких щелевидных дефектов. В подлежащих слоях межуточное вещество соединительной ткани подвергается мукоидному набуханию, ядра клеток лизируются, в то время как контуры пучков коллагеновых и мышечных клеток сохраняются.
 В некротизированных массах видны расширенные сосуды с гомогенными бесструктурными стенками, в просветах их нередко выявляются тромбы. Эти изменения мы обозначили как стадию некроза.
В некротизированных массах видны расширенные сосуды с гомогенными бесструктурными стенками, в просветах их нередко выявляются тромбы. Эти изменения мы обозначили как стадию некроза.Возможны два варианта заживления таких эрозий и язв.
В 27% наблюдений внутри или вокруг очагов некроза отмечалась выраженная лейкоцитарная инфильтрация с расплавлением некротических масс, их секвестрацией и отторжением (II стадия). В области краев или дна дефекта уже ко 2-м суткам можно увидеть новообразование капилляров и формирование грануляционной ткани. В биоптатах редко визуализируются все стадии процесса. Часто рядом с пластами МПЭ или изолированно обнаруживаются лишь очаговые скопления нейтрофилов, фибрина и/или некротических масс, указывающие на наличие острых пептических поражений.
Степень развития грануляционной ткани варьирует от очень мелких очагов в краях до обширных участков, занимающих всю площадь биоптата (рис. 1, б). Новообразованная молодая соединительная ткань представлена многочисленными сосудами капиллярного типа, иногда хорошо различимыми или невидимыми из-за обильной инфильтрации клеточными элементами. На поверхности грануляционной ткани видны скопления фибрина, лейкоцитов, а также микробов (чаще кокков), нити грибов типа Candida. При специальном окрашивании можно выявить характерный вертикальный ход капилляров к поверхности язвы. В глубоких отделах они располагаются параллельно поверхности дефекта и распространяются в мышечные слои. Эндотелий капилляров часто набухший, с крупными гиперхромными ядрами, на обычных препаратах приобретает сходство с опухолевыми клетками. Только использование специального окрашивания для выявления базальных мембран капилляров, а иногда и иммуногистохимических реакций позволяет выявить истинный характер изменений.
На поверхности грануляционной ткани видны скопления фибрина, лейкоцитов, а также микробов (чаще кокков), нити грибов типа Candida. При специальном окрашивании можно выявить характерный вертикальный ход капилляров к поверхности язвы. В глубоких отделах они располагаются параллельно поверхности дефекта и распространяются в мышечные слои. Эндотелий капилляров часто набухший, с крупными гиперхромными ядрами, на обычных препаратах приобретает сходство с опухолевыми клетками. Только использование специального окрашивания для выявления базальных мембран капилляров, а иногда и иммуногистохимических реакций позволяет выявить истинный характер изменений.
С момента повреждения начинается регенерация эпителиоцитов. В МПЭ происходят пролиферация клеток базального слоя (базально-клеточная гиперплазия), удлинение сосочков, что сопровождается значительным утолщением пласта; железистый эпителий в виде пласта смещается на эрозированную поверхность или грануляционную ткань. Нередко в эпителиоцитах желез, непосредственно граничащих с зоной повреждения, появляется смешанная желудочно-кишечная или кишечная слизь, интенсивно окрашивающаяся алциановым синим.
 В 74% наблюдений выявляется ЦМ кардиального типа: столбчатые клетки сходны с покровно-ямочным эпителием слизистой оболочки желудка, содержат в цитоплазме PAS+-слизь (рис. 1, д). В 26% наблюдений ЦМ расценена как кишечная. Во всех случаях она была неполной, сочеталась с ЦМ кардиального типа, имела разную распространенность (от отдельных бокаловидных клеток (БК) до крупных очагов с плоской или ворсинчатой поверхностью), наиболее обширной была в зоне СЦС, в краях глубоких язв (рис. 1, е). Помимо БК, в биоптатах обнаруживались и другие клеточные элементы кишечного типа: цилиндрические абсорбирующие с синей каймой, секретирующие с синей цитоплазмой, промежуточные клетки («тонко-толстокишечные», «желудочно-кишечные», «муцинозно-плоскоклеточные»). Количество БК возрастало с увеличением степени активности воспалительного процесса и снижалось при его затухании на фоне антисекреторной и противовоспалительной терапии. Таким образом формировался пищевод Барретта — участок ЦМ слизистой оболочки с кишечными бокаловидными и цилиндрическими клетками.
В 74% наблюдений выявляется ЦМ кардиального типа: столбчатые клетки сходны с покровно-ямочным эпителием слизистой оболочки желудка, содержат в цитоплазме PAS+-слизь (рис. 1, д). В 26% наблюдений ЦМ расценена как кишечная. Во всех случаях она была неполной, сочеталась с ЦМ кардиального типа, имела разную распространенность (от отдельных бокаловидных клеток (БК) до крупных очагов с плоской или ворсинчатой поверхностью), наиболее обширной была в зоне СЦС, в краях глубоких язв (рис. 1, е). Помимо БК, в биоптатах обнаруживались и другие клеточные элементы кишечного типа: цилиндрические абсорбирующие с синей каймой, секретирующие с синей цитоплазмой, промежуточные клетки («тонко-толстокишечные», «желудочно-кишечные», «муцинозно-плоскоклеточные»). Количество БК возрастало с увеличением степени активности воспалительного процесса и снижалось при его затухании на фоне антисекреторной и противовоспалительной терапии. Таким образом формировался пищевод Барретта — участок ЦМ слизистой оболочки с кишечными бокаловидными и цилиндрическими клетками.
При хронизации язв в глубоких отделах отмечается фиброзная ткань, содержащая коллагеновые волокна, фибробласты, лимфоидные и макрофагальные инфильтраты. На вертикальном срезе можно различить три слоя: фибринозно-лейкоцитарный (внутренний), грануляционной и фиброзной ткани с выраженной воспалительной инфильтрацией.
Помимо описанного варианта заживления пептических эрозий и язв возможен второй путь — без развития типичной грануляционной ткани, за счет организации некротических масс, который встречался в 15% наблюдений. Его развитию, на наш взгляд, способствует лейкоцитарно-макрофагальная недостаточность. Плотные гомогенные пикринофильные некротические массы не подвергаются расплавлению и отторжению лейкоцитами, а окружаются фиброзной тканью, инкапсулируются. Со стороны неизмененных тканей в них врастают немногочисленные сосуды и фибробласты, расслаивая на глыбки, которые в дальнейшем подвергаются гиалинозу и окрашиваются фуксином в красный цвет (рис. 2, а).Рисунок 2. Пептические эрозии и язвы слизистой оболочки пищевода при гастроэзофагеальной рефлюксной болезни. а — очаги гиалиноза в собственном слое слизистой оболочки при втором варианте заживления; б — пролиферация клеток многослойного плоского эпителия в краях каллезной язвы; в — острые эрозии слизистой оболочки на фоне цилиндроклеточной метаплазии кардиального типа; г — новообразование бокаловидных клеток при регенерации железистого эпителия. Окрашивание: а — пикрофуксином по Ван-Гизону; б, в — гематоксилином и эозином; г — PAS-реакция и алциановый синий. ×200. Эти гиалинизированные участки выявляются на дне эрозий и язв или сохраняются инкапсулированными в толще слизистой оболочки. Отсутствие характерной грануляционной ткани, богатой сосудами и клеточными элементами, являющимися источниками факторов роста, необходимых для регенерации эпителия, препятствует заживлению дефектов. На гиалинизированную соединительную ткань не наползает ни железистый, ни плоский эпителий. Так формируются своеобразные застывшие «каллезные» эрозии и язвы, с гиалинизированными краями и дном, иногда они частично покрыты резко уплощенным атрофированным эпителием.
Пептические эрозии и язвы слизистой оболочки пищевода при гастроэзофагеальной рефлюксной болезни. а — очаги гиалиноза в собственном слое слизистой оболочки при втором варианте заживления; б — пролиферация клеток многослойного плоского эпителия в краях каллезной язвы; в — острые эрозии слизистой оболочки на фоне цилиндроклеточной метаплазии кардиального типа; г — новообразование бокаловидных клеток при регенерации железистого эпителия. Окрашивание: а — пикрофуксином по Ван-Гизону; б, в — гематоксилином и эозином; г — PAS-реакция и алциановый синий. ×200. Эти гиалинизированные участки выявляются на дне эрозий и язв или сохраняются инкапсулированными в толще слизистой оболочки. Отсутствие характерной грануляционной ткани, богатой сосудами и клеточными элементами, являющимися источниками факторов роста, необходимых для регенерации эпителия, препятствует заживлению дефектов. На гиалинизированную соединительную ткань не наползает ни железистый, ни плоский эпителий. Так формируются своеобразные застывшие «каллезные» эрозии и язвы, с гиалинизированными краями и дном, иногда они частично покрыты резко уплощенным атрофированным эпителием.
Острые поражения на фоне предсуществующей ЦМ МПЭ СОП поверхностны (рис. 2, в). Это свидетельствует о том, что ЦМ является своеобразной приспособительной реакцией организма в ответ на воздействие кислого желудочного или щелочного дуоденального содержимого. Небольшие очаги некроза слизистой оболочки инфильтрируются нейтрофилами и отторгаются. При регенерации железистого эпителия появляются БК (рис. 2, г), усиливается новообразование желез. Многие из них кистозно расширяются. В их эпителиальной выстилке также появляются элементы с кишечной слизью и типичные БК. При каждом обострении число элементов с кишечной слизью увеличивается. Так расширяется зона пищевода Барретта.
При регенерации железистого эпителия появляются БК (рис. 2, г), усиливается новообразование желез. Многие из них кистозно расширяются. В их эпителиальной выстилке также появляются элементы с кишечной слизью и типичные БК. При каждом обострении число элементов с кишечной слизью увеличивается. Так расширяется зона пищевода Барретта.
Помимо пептических эрозий и язв, могут возникать деструктивные процессы другого происхождения. Так, при желудочной метаплазии слизистой оболочки часть эрозий связана с повреждением эпителия Helicobacter pylori. Такие изменения характеризуются поверхностным расположением, инфильтрацией нейтрофилами при отсутствии зон некроза. По сравнению с пептическими поражениями слабо выражена реактивная гиперплазия слизистых оболочек желез. Возможно участие и других микроорганизмов. Инфицирование микробами и грибами, как правило, сопровождается выраженным гнойным воспалением, новообразованием лимфоидных фолликулов, HPV и HSV — своеобразными изменениями многослойного плоского эпителия.
Конечной стадией репарации при острых эрозивно-язвенных поражениях является ремоделирование слизистой оболочки и подслизистого слоя в виде формирования на поверхности акантом и папиллом, железистых гиперплазиогенных полипов, репликации мышечной ткани t. muscularis mucosae, склероза, гиалиноза, ангиоматоза.
При развитии рубцовых стенозов в биоптатах выявляется резкое утолщение МПЭ с папилломатозом, гиперкератозом, акантозом, выраженным гиалинозом подлежащей соединительной ткани.
Таким образом, по данным биопсий, эрозивно-язвенные поражения СОП были выявлены в 41% наблюдений, несмотря на то, что эндоскопию и забор материала проводили обычно после курса терапии ИПП. Наиболее тяжелые эрозивно-язвенные формы наблюдаются в области СЦС или в зонах МПЭ. Можно выделить 5 типичных стадий их развития — некроз, лейкоцитарную инфильтрацию и отторжение некротических масс, гранулирование, эпителизацию и ремоделирование тканей после рубцевания. Заживление язв — длительный процесс, занимающий не менее 4—8 нед.
Помимо классического варианта нами описан второй возможный путь репарации — инкапсуляция и организация некротических масс, превращение их в очаги гиалиноза. В этих случаях слабо выражены лейкоцитарная и макрофагальная инфильтрация, резко угнетен ангиогенез, вследствие чего не формируется богато васкуляризированная соединительная (грануляционная) ткань. Нарушается реэпителизация, в краях дефектов отмечается активная пролиферация клеток многослойного плоского и железистого эпителия, в окружающих тканях происходит гиперплазия слизистых оболочек желез. Именно на этом фоне часто регистрируется интестинальная метаплазия и создаются условия для формирования предопухолевых состояний.
При предсуществующей ЦМ острые деструктивные поражения СОП поверхностны. В процессе регенерации эпителия происходит кишечная метаплазия покровного и железистого эпителия. Так постепенно формируется пищевод Барретта.
Следовательно, острые эрозии и язвы, возникающие в зонах многослойного плоского и цилиндрического эпителия, представляют собой закономерный этап в цепи изменений, предшествующих развитию пищевода Барретта и способствующих его прогрессированию.
Учитывая частоту пептических поражений СОП, длительность заживления, возникающие грубые морфологические изменения, в комплекс лечебных мероприятий помимо антисекреторной терапии следует, по-видимому, включать противовоспалительные и цитопротекторные препараты.
Все стадии развития пептических эрозий и язв должны найти отражение в гистологических заключениях по биоптатам СОП, и их необходимо учитывать при назначении местного и общего лечения.
COVID-19 и мужское репродуктивное здоровье
Фото носит иллюстративный характер. Из открытых источников
Прошел почти год, как мир впервые столкнулся с ранее неизвестным вирусным заболеванием, которое получило название COVID-19. Отдаленные последствия влияния вируса SARS-CoV-2 на организм человека на данный момент изучены недостаточно. При этом крайне важна оценка воздействия вируса на репродуктивное здоровье, чтобы своевременно предпринимать меры, способные предотвратить потерю фертильности и обеспечить сохранение демографической безопасности на мировом уровне.
Прямое повреждающее воздействие на ткань яичек
Светлана Жуковская, ассистент кафедры акушерства и гинекологии БГМУ
Накопленные клинические данные свидетельствуют, что вирус способен не только вызывать заболевания органов дыхательной системы, но и индуцировать патологические изменения в почках, печени, сердце и головном мозге. Также вирус SARS-CoV-2 был обнаружен в семенной жидкости у мужчин, больных COVID-19. Логично предположить, что, как и многие иные вирусы (корь, гепатит, простой герпес, ВИЧ), он способен вызывать повреждения ткани яичек, приводя тем самым к снижению фертильности.
Известно, что рецепторы к АПФ-2 (ангиотензин-превращающему ферменту 2) могут служить «точкой входа» в клетку для некоторых коронавирусов, в том числе для SARS-CoV и SARS-CoV-2. Рецепторы обнаруживаются во многих клетках человеческого организма (энтероциты, клетки почечных канальцев и желчного пузыря, кардиомиоциты, клетки мужских половых органов, трофобласт плаценты, эндотелиоциты и др. ), что объясняет широкий спектр патологических изменений при COVID-19.
), что объясняет широкий спектр патологических изменений при COVID-19.
Основные компоненты ренин-ангиотензин-альдостероновой системы (РААС) вовлечены в процесс регуляции стероидогенеза, сперматогенеза и обеспечения движения сперматозоидов в яичке и его придатке. В яичках и в мужском половом тракте обнаруживается высокая экспрессия АПФ-2; экспрессия матричной РНК АПФ-2 наблюдается как в герминативных, так и в соматических клетках яичка. В публикациях Z. Wang et al. (2020) и S. Verma et al. (2020) описана экспрессия АПФ-2 в сперматогониях, клетках Лейдига и Сертоли. В клетках Лейдига АПФ-2 участвует в регуляции синтеза стероидных гормонов, в эпителии семенных канальцев позволяет поддерживать процесс сперматогенеза. Отмечено, что уровень экспрессии АПФ-2 коррелирует с возрастом: экспрессия АПФ-2 начинается в период пубертата, достигая максимальной во время репродуктивного периода.
Установлено, что вирус SARS-CoV-2 может оказывать прямое повреждающее воздействие на ткань яичек: в публикации M. Yang et al. (2020) описаны результаты посмертного исследования тестикулярной ткани 12 пациентов с COVID-19 с использованием световой и электронной микроскопии. Выявлены такие изменения, как отек, вакуолизация, дилюция цитоплазмы в клетках Сертоли, разобщение базальной мембраны и семявыносящих канальцев, наличие клеточного детрита в просветах семявыносящих канальцев. Также отмечено значительное снижение количества клеток Лейдига в сравнении с контрольной группой, отек и воспалительная инфильтрация с преобладанием Т-лимфоцитов и гистиоцитов в интерстиции. Подтверждение повреждающего воздействия SARS-CoV-2 на тестикулярные клетки и ткани, а также на гематотестикулярный барьер отмечено в публикациях F. Chen et al. (2020), H. Ozveri et al. (2020) и K. Youssef et al. (2020), в которых описаны лейкоцитарная инфильтрация, нарушение сперматогенеза, распространенное повреждение герминативных клеток и резкое снижение количества либо полное отсутствие сперматозоидов в семенных канальцах, утолщение базальной мембраны, макрофагальная инфильтрация.
Yang et al. (2020) описаны результаты посмертного исследования тестикулярной ткани 12 пациентов с COVID-19 с использованием световой и электронной микроскопии. Выявлены такие изменения, как отек, вакуолизация, дилюция цитоплазмы в клетках Сертоли, разобщение базальной мембраны и семявыносящих канальцев, наличие клеточного детрита в просветах семявыносящих канальцев. Также отмечено значительное снижение количества клеток Лейдига в сравнении с контрольной группой, отек и воспалительная инфильтрация с преобладанием Т-лимфоцитов и гистиоцитов в интерстиции. Подтверждение повреждающего воздействия SARS-CoV-2 на тестикулярные клетки и ткани, а также на гематотестикулярный барьер отмечено в публикациях F. Chen et al. (2020), H. Ozveri et al. (2020) и K. Youssef et al. (2020), в которых описаны лейкоцитарная инфильтрация, нарушение сперматогенеза, распространенное повреждение герминативных клеток и резкое снижение количества либо полное отсутствие сперматозоидов в семенных канальцах, утолщение базальной мембраны, макрофагальная инфильтрация.
Влияние провоспалительных цитокинов
Известно, что SARS-CoV-2 может оказывать выраженное негативное воздействие на центральную нервную систему, проникая через гематоэнцефалический барьер и вызывая повреждение структур головного мозга. Поражение верхних уровней гипоталамо-гипофизарно-гонадной оси играет ключевую роль в нарушении фертильности, так как может приводить к гипогонадотропному гипогонадизму: дисфункция нейронов гипоталамуса, секретирующих гонадотропин-рилизинг-гормон, вызывает нарушение синтеза фолликулостимулирующего (ФСГ) и лютеинизирующего гормонов (ЛГ) клетками гипофиза, в результате чего нарушается функция клеток Сертоли и Лейдига. В публикации L. Ma et al. (2020) говорится о том, что у пациентов с COVID-19 наблюдалось значительное повышение концентрации ЛГ в сыворотке крови и снижение уровня ФСГ и тестостерона в сравнении с группой контроля, что оказало выраженное негативное влияние на сперматогенез.
Воздействие SARS-CoV-2 на репродуктивное здоровье мужчин также может быть опосредовано иммунными реакциями (цитокиновый шторм). Повышенное содержание провоспалительных цитокинов (ИЛ-6, ИЛ-1β, ФНО-α) оказывает негативное влияние на сперматогенез, приводя к нарушению фертильности. Цитокины вызывают супрессию гипоталамо-гипофизарно-гонадной оси: так, ИЛ-1 приводит к инактивации P450/c17-лиазы, которая обеспечивает конверсию прогестинов в андрогены, что проявляется снижением тестостерона и нарушением сперматогенеза. Повышение экспрессии ИЛ-6 приводит к нарушению целостности гематотестикулярного барьера, позволяя вирусу оказывать прямое повреждающее воздействие на ткань яичек.
Повышенное содержание провоспалительных цитокинов (ИЛ-6, ИЛ-1β, ФНО-α) оказывает негативное влияние на сперматогенез, приводя к нарушению фертильности. Цитокины вызывают супрессию гипоталамо-гипофизарно-гонадной оси: так, ИЛ-1 приводит к инактивации P450/c17-лиазы, которая обеспечивает конверсию прогестинов в андрогены, что проявляется снижением тестостерона и нарушением сперматогенеза. Повышение экспрессии ИЛ-6 приводит к нарушению целостности гематотестикулярного барьера, позволяя вирусу оказывать прямое повреждающее воздействие на ткань яичек.
В контексте оценки влияния заболевания COVID-19 на мужское репродуктивное здоровье следует помнить о том, что нормальное развитие сперматозоидов возможно при температуре, не превышающей 37,8 °C. Выраженная гипертермия, как локальная, так и системная, приводит к снижению количества сперматозоидов, нарушению их подвижности, возникновению анеуплоидий.
Влияние глюкокортикоидов
Отдельно следует отметить, что глюкокортикоиды, нередко применяемые для лечения COVID-19, оказывают прямое негативное влияние на сперматогенез: в экспериментальном исследовании установлено, что дексаметазон ингибирует дифференциацию клеток Лейдига, снижая экспрессию Cyp17a1 и Scarb1.
Тревога увеличивает ДНК-фрагментацию
Исследователи указывают на важность психологического состояния пациентов с COVID-19: высокий уровень тревоги, стресс и депрессивные состояния приводят к снижению синтеза секс-связывающего глобулина, повышению уровня кортизола и пролактина, что в свою очередь ведет к снижению количества и концентрации сперматозоидов, возрастанию процента ДНК-фрагментации и к расстройствам эрекции и эякуляции.
Очевидно, что сохранение мужского репродуктивного здоровья во время пандемии COVID-19 является актуальной проблемой современной медицины, которая требует дальнейшего углубленного изучения.
Мужчинам, перенесшим COVID-19 и планирующим отцовство, целесообразно рекомендовать проведение преконцепционного обследования: спермограмма со строгой оценкой морфологии, MAR-тест и исследование ДНК-фрагментации сперматозоидов.
Случаи орхита
Некоторые исследования описывают орхит, индуцированный вирусом SARS-CoV-2. По данным F. Pan et al. (2020), у 19 % мужчин с COVID-19 отмечался дискомфорт в области мошонки; в публикации H. Ozveri et al. (2020) описано умеренное увеличение васкуляризации яичек при проведении ультразвукового исследования у пациента с COVID-19, предъявлявшего жалобы на ощущение распирания и боли в области мошонки, однако изменений в показателях спермограммы у этого пациента отмечено не было. Воспаление, ассоциированное с SARS-CoV-2, описано в публикации L. Gagliardi et al. (2020), в которой представлен клинический случай орхоэпидидимита у 14-летнего мальчика, больного COVID-19. В работе M. Pouletty et al. (2020) также описаны 2 случая орхита у детей на фоне системного воспалительного синдрома, вызванного COVID-19.
По данным F. Pan et al. (2020), у 19 % мужчин с COVID-19 отмечался дискомфорт в области мошонки; в публикации H. Ozveri et al. (2020) описано умеренное увеличение васкуляризации яичек при проведении ультразвукового исследования у пациента с COVID-19, предъявлявшего жалобы на ощущение распирания и боли в области мошонки, однако изменений в показателях спермограммы у этого пациента отмечено не было. Воспаление, ассоциированное с SARS-CoV-2, описано в публикации L. Gagliardi et al. (2020), в которой представлен клинический случай орхоэпидидимита у 14-летнего мальчика, больного COVID-19. В работе M. Pouletty et al. (2020) также описаны 2 случая орхита у детей на фоне системного воспалительного синдрома, вызванного COVID-19.
Светлана Жуковская, ассистент кафедры акушерства и гинекологии БГМУ
Медицинский вестник, 28 декабря 2020
Поделитесь
Кальцификация как патогенетическая основа атеросклероза
Сафошкина Е. В.
В.
Процесс кальцификации характеризуется отложением фосфатов кальция (гидроксиапатитов, оксалатов кальция и октакальциевых фосфатов) в мягких тканях: в стенке кровеносных сосудов, миокарде, клапанном аппарате сердца, а также в почечной ткани, коже, сухожилиях [1]. Чрезвычайно опасный для жизни и весьма распространенный вид кальциноза связан с локализацией отложений фосфатов кальция в артериях и клапанах сердца. Этот вид патологии приводит к тяжелой патологии и высокой летальности [1]. Несмотря на многочисленные и всесторонние исследования клеточного и биохимического механизмов кальцификации, многие проблемы в этой области остаются невыясненными. Кальцификация коронарных сосудов встречается у большинства пациентов с ишемической болезнью сердца, а также у пациентов с разрывами крупных сосудов. В артериях кальцификация сочетается с формированием атеросклеротических бляшек, что увеличивает риск инфарктов, частоту развития эпизодов ишемии. Эти процессы в сосудах, пораженных атеросклерозом, ведут к еще большему повреждению их стенки, а также к повышению хрупкости стенки сосуда, что связано с риском разрыва атеросклеротической бляшки, тромбозом сосудов, а также расслоением сосудистой стенки [4]. Кальцификация коронарных артерий усугубляет ишемические процессы в миокарде, тем самым значительно повышая риск неблагоприятных сердечно-сосудистых событий и смертности.
Кальцификация коронарных артерий усугубляет ишемические процессы в миокарде, тем самым значительно повышая риск неблагоприятных сердечно-сосудистых событий и смертности.
Первые попытки феноменологического объяснения наблюдаемых процессов привели к концепции, согласно которой кальцификация мягких тканей является дегенеративным процессом, приводящим к патологическому отложению фосфатов кальция в мягких тканях [3]. В последние годы общие представления о процессе кальцификации мягких тканей радикально изменились. В настоящее время принята концепция, согласно которой кальцификация мягких тканей является патологическим, регулируемым процессом, в котором участвуют клетки, а также биохимическая система, которая осуществляет регуляцию [3]. Иными словами, эктопическая кальцификация является сложным регулируемым клеточно-биохимическим процессом. Эта концепция открывает перспективу создания методов профилактики, а также, возможно, реверсирования процесса кальцификации [5].
В более поздних исследованиях, в частности, в обзоре L. Demer процессы кальцификации сосудистой стенки и клапанного аппарата рассматриваются в общем ключе процесса атеросклероза [10]. В частности, в кальцифицированном аортальном клапане человека происходит перемещение эластина, накопление липидов, воспалительные процессы, которые ведут к усилению процессов кальцификации. Существенным отличием позиции автора от других является предположение о наличии остеохондрогенной дифференцировки клеток клапанного аппарата вкупе с взаимным влиянием на процессы атеросклероза.
Demer процессы кальцификации сосудистой стенки и клапанного аппарата рассматриваются в общем ключе процесса атеросклероза [10]. В частности, в кальцифицированном аортальном клапане человека происходит перемещение эластина, накопление липидов, воспалительные процессы, которые ведут к усилению процессов кальцификации. Существенным отличием позиции автора от других является предположение о наличии остеохондрогенной дифференцировки клеток клапанного аппарата вкупе с взаимным влиянием на процессы атеросклероза.
Согласно предлагаемой концепции, при повреждении атеросклерозом сосудистой стенки наблюдается нарушение дифференцировки сосудистой ткани и, следовательно, репаративных процессов. Дифференцировка клеточных элементов сосудистой ткани происходит в неполной степени, т.е. вместо зрелых клеток в поврежденную сосудистую ткань “приходят” менее дифференцированные предшественники, которые обладают свойствами клеток костной ткани [2]. Кроме того, мультипотентные мезенхимальные клетки, чувствительные к факторам роста, находятся как в участках ангиогенеза, так и в участках атеросклеротического повреждения артерий. Воспаление и макрофагальная инфильтрация усиливает остеогенную активность в очаге атеросклероза. Кристаллы кальция, взаимодействуя с макрофагами, усиливают выработку последними провоспалительных цитокинов, поддерживая положительную обратную связь между кальцификацией и прогрессированием воспаления. В некоторых случаях в такие участки формирования сосудистой ткани вовлекаются и “другие клетки”, в частности, предшественники нервной ткани. В некоторых публикациях показаны существенные различия в субпопуляциях клеток сосудистой ткани (в частности, их ответ на воздействие факторов роста и способность к дифференцировке) [8,9].
Воспаление и макрофагальная инфильтрация усиливает остеогенную активность в очаге атеросклероза. Кристаллы кальция, взаимодействуя с макрофагами, усиливают выработку последними провоспалительных цитокинов, поддерживая положительную обратную связь между кальцификацией и прогрессированием воспаления. В некоторых случаях в такие участки формирования сосудистой ткани вовлекаются и “другие клетки”, в частности, предшественники нервной ткани. В некоторых публикациях показаны существенные различия в субпопуляциях клеток сосудистой ткани (в частности, их ответ на воздействие факторов роста и способность к дифференцировке) [8,9].
Повреждение сосудистой стенки с последующим нарушением дифференцировки клеточных элементов, вероятно, является составным компонентом в кальцификации сосудистой ткани. Это наглядно демонстрирует исследование на животных на примере эктопической кальцификации у крыс, у которых имелись атеросклеротические бляшки [7].
Во многих международных многоцентровых клинических исследованиях показано влияние липидснижающих препаратов на подавление клапанной кальцификации [11]. Антикальцификационное действие статинов находит объяснение в снижении уровня ЛПНП-ассоциированного белка (LRP5) – независимого активатора гена Wnt, включающим аккумуляцию в ядерном аппарате кардиомиоцита β-катенина, который является необходимым элементом для развития остеобластогенеза. Кроме того, в пользу указанного механизма свидетельствует эмбриональное происхождение аортального калапана из нервной трубки, указанный процесс также контролируется геном Wnt. L. Demer, Y. Tintut и K. Bostrom в своих обзорах указывают на возможность формирования клеток с характеристиками остеобластов из мезенхимальной ткани под воздействием различным факторов роста. Указанные процессы имеют большую вероятность развития в поврежденных атеросклерозом гладкомышечных клетках эндотелия, а также клапанном аппарате сердца [12]. Кроме того, процессы оссификации гистологическими методами обнаружены примерно у 60% пациентов после баллонной ангиопластики по поводу рестеноза аортального отверстия; примерно в 15% случаев атеросклеротические бляшки в сонных артериях и клапанном аппарате сердца имели признаки оссификации [2].
Антикальцификационное действие статинов находит объяснение в снижении уровня ЛПНП-ассоциированного белка (LRP5) – независимого активатора гена Wnt, включающим аккумуляцию в ядерном аппарате кардиомиоцита β-катенина, который является необходимым элементом для развития остеобластогенеза. Кроме того, в пользу указанного механизма свидетельствует эмбриональное происхождение аортального калапана из нервной трубки, указанный процесс также контролируется геном Wnt. L. Demer, Y. Tintut и K. Bostrom в своих обзорах указывают на возможность формирования клеток с характеристиками остеобластов из мезенхимальной ткани под воздействием различным факторов роста. Указанные процессы имеют большую вероятность развития в поврежденных атеросклерозом гладкомышечных клетках эндотелия, а также клапанном аппарате сердца [12]. Кроме того, процессы оссификации гистологическими методами обнаружены примерно у 60% пациентов после баллонной ангиопластики по поводу рестеноза аортального отверстия; примерно в 15% случаев атеросклеротические бляшки в сонных артериях и клапанном аппарате сердца имели признаки оссификации [2]. Гистологическими исследованиями подтверждено, что структуры, сходные с костной тканью, существуют внутри атеросклеротических образований, причем процессы, протекающие в них, сходны с процессами ремоделирования в обычной трабекулярной кости [6]. Несмотря на то, что механизмы кальцификации и атеросклероза перекликаются между собой, не всегда кальцификация протекает в присутствии атеросклеротических изменений [2]. Метаболические нарушения такие как сахарный диабет, уремия, гиперпаратиреоидизм, а также артериальная гипертония, связаны с процессами кальцификации, которые протекают даже в отсутствии атеросклероза [13].
Гистологическими исследованиями подтверждено, что структуры, сходные с костной тканью, существуют внутри атеросклеротических образований, причем процессы, протекающие в них, сходны с процессами ремоделирования в обычной трабекулярной кости [6]. Несмотря на то, что механизмы кальцификации и атеросклероза перекликаются между собой, не всегда кальцификация протекает в присутствии атеросклеротических изменений [2]. Метаболические нарушения такие как сахарный диабет, уремия, гиперпаратиреоидизм, а также артериальная гипертония, связаны с процессами кальцификации, которые протекают даже в отсутствии атеросклероза [13].
Примером, демонстрирующим сложность процессов про- и антикальцификации, многообразие регулирующих систем, работающих в равновесии друг с другом, может служить следующий клинический случай, к сожалению, не являющийся редкостью в повседневной практике врача-клинициста. При первичном обращении в декабре 2009 года у пациента К., 1927 года рождения, около 40 лет страдающего сахарным диабетом, компенсированного диетой, ишемической болезнью сердца (стенокардией напряжения, ФК I), пароксизмальной формой мерцательной аритмии, были отмечены гипергликемия натощак 10,3 ммоль/л, гиперхолестеринемия 7,7 ммоль/л, эхокардиографические признаки атеросклеротических изменений восходящего отдела аорты, кальциноз фиброзного кольца, гипертрофия левого желудочка с формированием сочетанного порока аортального клапана (умеренного стеноза устья аорты, недостаточности АК I-II степени), кальциноз створок АК, недостаточность МК II степени, в бульбусе с переходом на устье правой внутренней сонной артерии на передней стенке гиперэхогенная бляшка с нарушением целостности покрышки длиной 2 см, стенозировавшая просвет слева до 60%, справа в общей сонной артерии – стабильная бляшка с кальцинозом и стеноз 40-45%.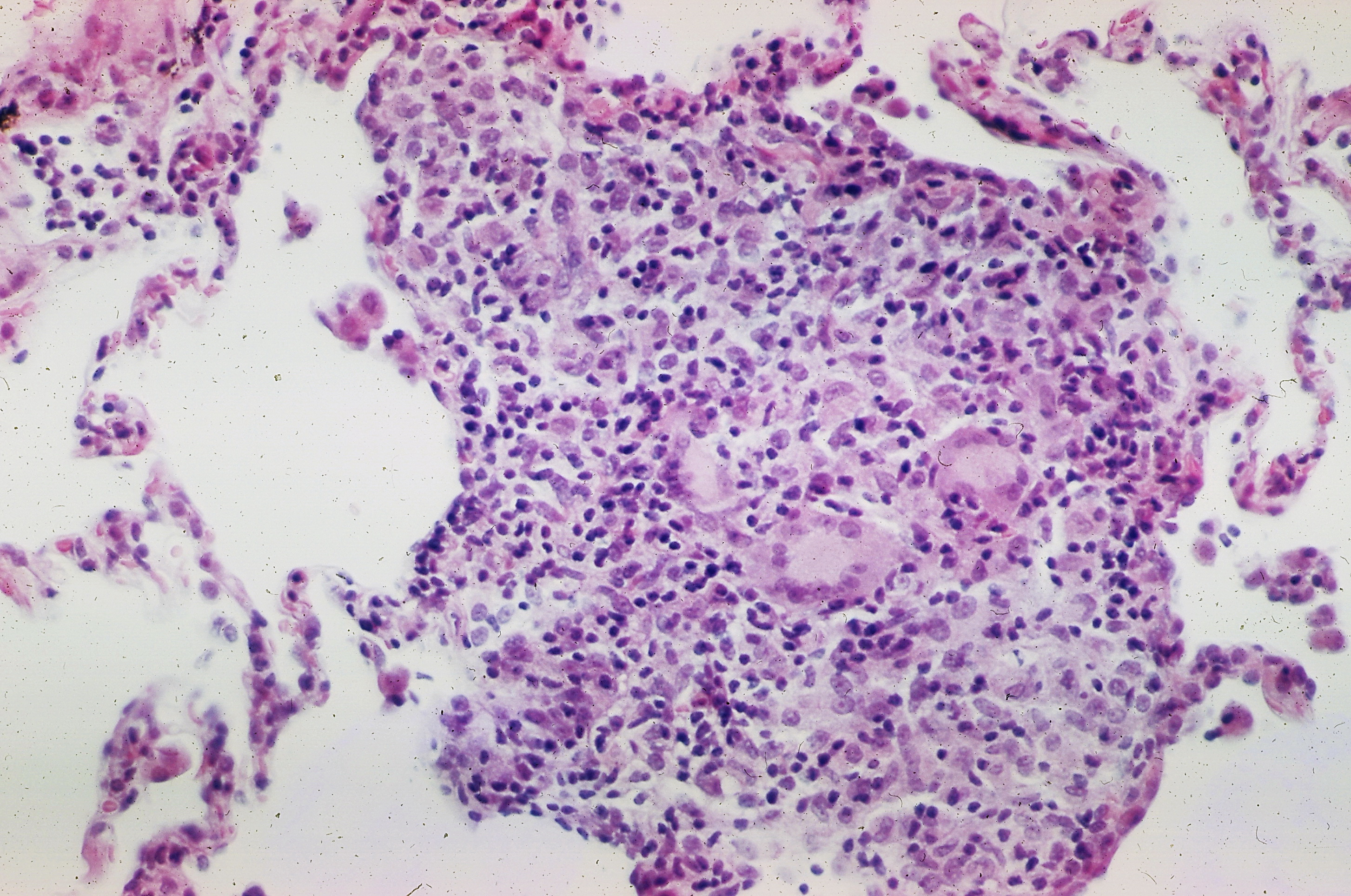 С учетом дообследования было назначено следующее лечение: конкор 5 мг в сутки, диабетон 60 мг в сутки, крестор 10 мг в сутки с последующим динамическим наблюдением в течение 2-х лет. За это время удалось стабилизировать уровень гликемии до 4,8-5,3 ммоль/л, показатели общего холестерина были снижены до 3,5 ммоль/л. Контрольная эхокардиография не отметила дальнейшего прогрессирования кальциноза клапанного аппарата сердца. По данным ультрасонографии с допплер-анализом произошла стабилизация атеросклеротической бляшки левой сонной артерии, кроме того, зафиксировано уменьшение стеноза – слева до 40%, справа до 25-30%.
С учетом дообследования было назначено следующее лечение: конкор 5 мг в сутки, диабетон 60 мг в сутки, крестор 10 мг в сутки с последующим динамическим наблюдением в течение 2-х лет. За это время удалось стабилизировать уровень гликемии до 4,8-5,3 ммоль/л, показатели общего холестерина были снижены до 3,5 ммоль/л. Контрольная эхокардиография не отметила дальнейшего прогрессирования кальциноза клапанного аппарата сердца. По данным ультрасонографии с допплер-анализом произошла стабилизация атеросклеротической бляшки левой сонной артерии, кроме того, зафиксировано уменьшение стеноза – слева до 40%, справа до 25-30%.
Безусловно, процессы кальцификации пока еще не до конца изучены. Рассмотренные выше механизмы могут являться ключом к разгадке этой проблемы. Появление возможности количественного определения некоторых медиаторов кальцификации, очевидно, будет способствовать более точному определению времени наступления кальцификации. Это в свою очередь позволит значительно расширить возможности превентивной терапии с учетом необходимых потребностей, пересмотреть лекарственную терапию в дополнение к уже используемым препаратам, а также создать новые, более эффективные с учетом современных представлений о процессах кальцификации в организме.
Список литературы:
- Giachelli C.M. Ectopic calcification: gathering hard facts about soft tissue mineralization. Am J Path.,1999;3;671-675.
- Abedin M., Tintut Y., Demer L.L. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 2004;24;1161-1170.
- Cotran RS, Kumare V, Robbins SL Cellular injury and cellular death. Patho- logical Basis of Disease, 5th ed.,1994, 1-35.
- Stein GS, Lian JB. Molecular mechanisms mediating developmental and hormoneregulated expression of genes in osteoblasts: an integrated relationship of cell growth and differentiation. Cellular and Molecular Biology of Bone, 1993; 48-95.
- Boström K. Cell differentiation in vascular calcification. Z Kardiol 2000; 89(suppl 2): II-69-II-74.
- Jeziorska M, McCollum C,Wooley DE. Observations on bone formation and remodeling in advanced atherosclerotic lesions of human carotid arteries. Virchows Arch. 1998;433;559-565.

- Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilages in mice lacking matrix GLA protein. Nature. 1997; 386; 78-81.
- Hungerford JE, Little CD. Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J Vasc Res 1999;36;2-27.
- Owens G. Regulation of differention of vascular smooth muscle cells. Phys Rev; 1995; 75; 487-517.
- Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008 Jun 3;117(22):2938-48.
- Rajamannan N.M., Subramaniam M., Stock S.R., Stone N.J., Springett M., Ignatiev K.I., McConnell J.P., Singh R.J., Bonow R.O., Spelsberg T.C. Atorvastatin inhibits calcification and enhances duction in the hypercholesterolaemic aort. Heart. 2005, 91:806-810.
- Boström K. Insights into the mechanism of vascular calcification. Am J Cardiol. 2001 Jul 19;88(2A):20E-22E.
- Murshed M, Harmey D, Millán JL, McKee MD, Karsenty G. Unique coex- pression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005 May 1;19(9):1093-104. Epub 2005 Apr 15.

 Unique coex- pression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005 May 1;19(9):1093-104. Epub 2005 Apr 15.
Unique coex- pression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005 May 1;19(9):1093-104. Epub 2005 Apr 15.Дифференциальная макрофагальная инфильтрация при раннем и распространенном эндометриозе и прилежащей брюшине
Цель: Исследовать распределение макрофагальной (Mphi) инфильтрации в эутопическом и эктопическом эндометрии на протяжении менструального цикла.
Дизайн: Контролируемое клиническое исследование с использованием интактной ткани.
Параметр: Медицинский факультет Университета Нагасаки, Нагасаки, Япония.
Пациент(ы): Двадцать бесплодных женщин с тазовым эндометриозом и 20 женщин без эндометриоза.
Вмешательство(я): Образцы биопсии из поражений брюшины и соответствующего эутопического эндометрия были взяты у женщин с эндометриозом или без него.Прилежащие перитонеальные биопсии также были получены у части этих женщин. Активированный маркер Mphi CD68, митогенный маркер фактора роста гепатоцитов (HGF) и маркер поверхности эндотелиальных клеток фактор фон Виллебранда были иммунолокализованы и количественно оценены с помощью световой микроскопии и оценки Q-H.
Основные показатели результата: Исследовали тканевую инфильтрацию Mphi в эутопическом эндометрии, эктопическом эндометрии и прилежащей брюшине и анализировали ее взаимосвязь с иммунореакцией HGF и количеством микрососудов.Также исследовали возможную продукцию HGF изолированным базальным Mphi.
Результаты): Тканевая инфильтрация Mphi в эутопическом и эктопическом эндометрии женщин с эндометриозом I-II стадии была значительно выше, чем с эндометриозом III-IV стадии или у женщин контрольной группы. Красные поражения брюшины и прилегающая к ним брюшина имели наибольшую концентрацию Mphi по сравнению с черными или белыми поражениями.Эти воспалительные клетки показали более высокое распространение в секреторной фазе менструального цикла. Плотность MPHI в эутопическом эндометрии и соответствующих красных поражениях показала значительную корреляцию как с оценкой Q-H HGF, так и с плотностью микрососудов. Значительное количество HGF также продуцируется изолированным базальным Mphi от женщин с эндометриозом.
Вывод(ы): Эти результаты позволяют предположить, что перитонеальные поражения раннего и активного эндометриоза и прилегающая к ним брюшина содержат большое количество Mphi, которые могут быть вовлечены в рост эндометриоза.
Дисрегуляция опухолеассоциированных макрофагов в канцерогенезе, прогрессировании и таргетной терапии гинекологического рака и рака молочной железы | Journal of Hematology & Oncology
Согласно глобальной статистике рака за 2020 год, на общее количество гинекологических опухолей, включая рак шейки матки, вульвы, тела матки, яичников и влагалища, приходится до 15% общей новой заболеваемости женщин во всем мире. Кроме того, на рак молочной железы также приходится около четверти всех новых случаев рака и каждый шестой случай смерти от рака у женщин [1].Хотя комбинированная хирургия с химиолучевой терапией или без нее, иммунотерапией и таргетной терапией значительно улучшила клинические исходы у пациентов с гинекологическим раком и раком молочной железы, общая выживаемость при запущенных заболеваниях все еще низка.
В последние годы новая концепция и область изучения микроокружения опухоли (TME) стали горячей темой в исследованиях рака. Опухолевые клетки окружены динамической средой, которая включает макрофаги, лимфоциты, мезенхимальные стволовые клетки, фибробласты, а также другие иммунные, воспалительные и стромальные клетки и различные биохимические молекулы.Среди этих компонентов ТМЭ опухолеассоциированные макрофаги (ТАМ) являются основными действующими лицами, на долю которых приходится от 30 до 50% клеток ТМЭ [2], которые играют решающую роль в ангиогенезе, метастазировании опухоли, иммуносупрессии и терапевтической резистентности [3]. ]. Интересно, что гинекологические опухоли имеют разные перекрестные помехи с ТАМ из-за их характеристик. Например, персистирующая инфекция вирусом папилломы человека (ВПЧ) высокого риска является одной из основных причин рака шейки матки. Онкобелки HPV E6/E7 не только изменяют клеточную пролиферацию и интерфероновый ответ, воздействуя на экспрессию цитокинов [4, 5], но также имеют многочисленные ассоциации с ТАМ вместе, способствуя развитию опухоли [2].Кроме того, с ТАМ взаимодействуют рецепторы эстрогенов, которые в высокой степени экспрессируются при раке эндометрия, яичников и молочной железы, способствуя миграции и инвазии раковых клеток [6, 7].
Опухолевые клетки окружены динамической средой, которая включает макрофаги, лимфоциты, мезенхимальные стволовые клетки, фибробласты, а также другие иммунные, воспалительные и стромальные клетки и различные биохимические молекулы.Среди этих компонентов ТМЭ опухолеассоциированные макрофаги (ТАМ) являются основными действующими лицами, на долю которых приходится от 30 до 50% клеток ТМЭ [2], которые играют решающую роль в ангиогенезе, метастазировании опухоли, иммуносупрессии и терапевтической резистентности [3]. ]. Интересно, что гинекологические опухоли имеют разные перекрестные помехи с ТАМ из-за их характеристик. Например, персистирующая инфекция вирусом папилломы человека (ВПЧ) высокого риска является одной из основных причин рака шейки матки. Онкобелки HPV E6/E7 не только изменяют клеточную пролиферацию и интерфероновый ответ, воздействуя на экспрессию цитокинов [4, 5], но также имеют многочисленные ассоциации с ТАМ вместе, способствуя развитию опухоли [2].Кроме того, с ТАМ взаимодействуют рецепторы эстрогенов, которые в высокой степени экспрессируются при раке эндометрия, яичников и молочной железы, способствуя миграции и инвазии раковых клеток [6, 7]./108/108.jpg) В наших предыдущих исследованиях мы также обнаружили корреляцию между инфильтрацией ТАМ и гинекологическим раком [8,9,10].
В наших предыдущих исследованиях мы также обнаружили корреляцию между инфильтрацией ТАМ и гинекологическим раком [8,9,10].
Поэтому в этом обзоре мы сосредоточимся на ТАМ при гинекологическом раке и раке молочной железы с акцентом на их происхождение, функцию и регуляцию опухолевой прогрессии и метастазирования. Мы также рассматриваем многообещающие таргетные терапии ТАМ для ингибирования проопухолевых ТАМ и новые иммунотерапевтические средства на основе ТАМ, включая макрофаги CAR, вакцинацию и нанобиотехнологии, которые находятся в полном расцвете.
Происхождение и развитие ТАМ
Являясь жизненно важным компонентом врожденной иммунной системы, макрофаги принадлежат к мононуклеарной фагоцитарной системе, которая может поглощать патогены, ингибировать воспаление и координировать восстановление тканей [11, 12]. Макрофаги в тканях состоят из двух частей: резидентных макрофагов желточного мешка (TRM) и моноцитов крови из костного мозга [13]. TAM представляют собой макрофаги, собранные в TME [14], и большинство TAM представляют собой макрофаги костного мозга (BMDM), рекрутируемые хемотаксическими и цитокинами, такими как моноцитарный хемотаксический белок 1 (MCP-1/CCL2) и колониестимулирующий фактор 1 ( КСФ-1) [3]. Как правило, гемопоэтические стволовые клетки (ГСК) дифференцируются в клетки-предшественники костного мозга (КМП), затем проходят через ограниченные гранулоцитами/макрофагами клетки-предшественники (ГМП) и клетки-предшественники макрофагов/дендритов (МДП) и обладают способностью дифференцироваться в моноциты и мононуклеарные миелоидные супрессорные клетки (M-MDSC) [15]. Как только эти клетки попадают в ТМО, локальные индукторы постепенно регулируют дифференцировку моноцитов в макрофаги проопухолевого или противоопухолевого типа (таблица 1) [16,17,18,19,20,21,22,23,24, 25,26,27,28,29,30,31].ТАМ различного происхождения можно отличить по экспрессии поверхностных маркеров (таких как CX3CR1, CD45, CCR2, MERTK и FcRγ1/CD64), профилю экспрессии генов и трекерам клонов (рис. 1) [13]. Кроме того, были обнаружены различные функции ТАМ, например,
TRM связаны с пролиферацией клеток и фиброзом, тогда как BMDM связаны с презентацией антигена [32], которая в некоторой степени влияет на рост и метастазирование опухолевых клеток и обеспечивает направления исследований для возможных стратегий лечения рака [33, 35, 36].
Как правило, гемопоэтические стволовые клетки (ГСК) дифференцируются в клетки-предшественники костного мозга (КМП), затем проходят через ограниченные гранулоцитами/макрофагами клетки-предшественники (ГМП) и клетки-предшественники макрофагов/дендритов (МДП) и обладают способностью дифференцироваться в моноциты и мононуклеарные миелоидные супрессорные клетки (M-MDSC) [15]. Как только эти клетки попадают в ТМО, локальные индукторы постепенно регулируют дифференцировку моноцитов в макрофаги проопухолевого или противоопухолевого типа (таблица 1) [16,17,18,19,20,21,22,23,24, 25,26,27,28,29,30,31].ТАМ различного происхождения можно отличить по экспрессии поверхностных маркеров (таких как CX3CR1, CD45, CCR2, MERTK и FcRγ1/CD64), профилю экспрессии генов и трекерам клонов (рис. 1) [13]. Кроме того, были обнаружены различные функции ТАМ, например,
TRM связаны с пролиферацией клеток и фиброзом, тогда как BMDM связаны с презентацией антигена [32], которая в некоторой степени влияет на рост и метастазирование опухолевых клеток и обеспечивает направления исследований для возможных стратегий лечения рака [33, 35, 36].
Поверхностные маркеры ТРМ и МДМ
Поляризация ТАМ
По разным факторам поляризации и иммунным функциям макрофаги можно разделить на два типа: а именно классически активированные М1 и альтернативно активированные макрофаги М2 [37]. Макрофаги M1 в основном индуцируются интерфероном γ (IFNγ), липополисахаридом (LPS) и гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF) и оказывают цитотоксическое действие на раковые клетки.Напротив, макрофаги М2 могут обеспечить питательное преимущество раковым клеткам при стимуляции ИЛ-4, ИЛ-6, ИЛ-10, ИЛ-13, трансформирующим фактором роста β (TGF-β), фактором роста эндотелия сосудов (VEGF) и другие М2-специфические цитокины [34, 38, 39]. Обычно предполагается, что ТАМ в значительной степени поляризованы в макрофаги М2, что способствует прогрессированию опухоли и сдерживанию противоопухолевого иммунного ответа.
Эта тенденция к поляризации может быть связана с различными индуцируемыми факторами, секретируемыми составляющими клетками в TME (таблица 1).Например, Pedraza-Brindis et al. сообщили, что супернатант клеток рака шейки матки подавлял фосфорилирование NF-κB-p65 и STAT1 в макрофагах M1 и индуцировал стабильный фенотип M2, сопровождающийся повышенной экспрессией IL-10, IL-6, CCL2, IL-8, GM-CSF, G-CSF, фактор роста тромбоцитов (PDGF) AA и PDGF-BB [40, 41]. Эксперименты показали, что клетки рака шейки матки секретируют простагландин (PG) E2 и IL-6, чтобы индуцировать фенотип M2, и продуцируют CCL2, чтобы способствовать рекрутированию моноцитов в месте опухоли [34].Более того, CSF-1 может индуцировать поляризацию М2 в клетках рака яичников с прогрессированием гистологической злокачественности [42]. Кроме того, блокирование рецептора CSF-1 (CSF-1R) может предотвратить инфильтрацию макрофагов и пролиферацию раковых клеток эндометрия [43]. Более того, лечение паклитакселом клеток рака молочной железы усиливает секрецию CSF-1, рекрутируя ТАМ, чтобы ограничить терапевтический эффект [44].
Влияние бескислородного микроокружения на ТАМ
Быстрая пролиферация опухолевых клеток приводит к недостаточности кровотока, накоплению молочной кислоты и гипоксии при ТМЭ, что приводит к усилению ангиогенеза и катализирует рост опухоли [49].В TME с низкой концентрацией кислорода опухолевые клетки могут рекрутировать ТАМ, секретируя больше хемокинов, таких как CCL2, CCL5 или CSF1 [3]. Было подтверждено, что при ТМЭ рака шейки матки гипоксическое состояние активирует М2-подобные ТАМ in vivo [45]. Одно предыдущее исследование показало, что макрофаги индуцировались в гипоксических областях опухоли и приобретали M2-поляризованный фенотип с помощью цитокинов, полученных из гипоксических раковых клеток, онкостатина М (OSM), воспалительного цитокина, принадлежащего к суперсемейству IL-6. Недавно было доказано, что это явление опосредовано сигнальным комплексом 2 mTOR (mTORC2) [46, 47].Кроме того, исследования экзосом показали, что рак эндометрия с гипоксией опосредует поляризацию моноцитов в макрофаги М2 посредством экссудации экзокринной миРНК-21 [48]. Между тем, гипоксическая ТМЭ также приводит к снижению подвижности внеклеточного матрикса и более медленному движению ТАМ, что усиливает взаимодействие между ТАМ и раковыми клетками [50].
Между тем, гипоксическая ТМЭ также приводит к снижению подвижности внеклеточного матрикса и более медленному движению ТАМ, что усиливает взаимодействие между ТАМ и раковыми клетками [50].
Были также проведены эксперименты, чтобы продемонстрировать, играет ли индуцируемый гипоксией ZEB1, ключевой регулятор эпителиально-мезенхимальной трансформации (ЭМП), роль в рекрутировании ТАМ при гипоксическом раке шейки матки TME.Результаты показали, что ZEB1 активировал CCL8, который в сочетании с рецептором CCL8 (CCR2) и дополнительно рекрутировал ТАМ посредством сигнального фосфорилирования NF-κB [51].
Биомаркеры ТАМ
Маркеры клеточной поверхности представляют собой класс веществ, обычно белков, которые экспрессируются на поверхности клеток для идентификации типов или свойств клеток. На поверхности макрофагов имеется много видов рецепторов, среди которых широко изучены рецепторные тирозинкиназы Tyro3, Axl и MerTK [52].Когда эти рецепторы соединяются с лигандами, такими как специфичность ингибирования роста 6 (Gas6) и белок S, макрофаги поощряются к поляризации на M2 посредством проведения специфических путей, среди которых передача сигналов PI3K/Akt является наиболее изученным путем [53,54]. ,55].
,55].
За исключением рецепторов на макрофагах, эти клетки также экспрессируют MHC-II и CD на цитомембране для участия в иммунном ответе. В целом, макрофаги M1 экспрессируют CD14, CD16/33, CD40, CD80 и CD86, продуцируют IL-6, IL-12p70 и IL-23 и играют провоспалительную, цитотоксическую и тумороцидную роль.Что касается макрофагов М2, то в качестве маркеров идентифицированы аргиназа 1 (Arg-1), CD163, CD169, CD206, IL-10 и хемокины CCL17 и CCL22 [57, 58]. Панмакрофагальные маркеры также участвуют в обнаружении клеток макрофагов, таких как CD68, CD14, CD45, CD105 и CD204 [14, 40]. Много лет назад в некоторых публикациях ТАМ просто включались в М2-подобные макрофаги [56], что сегодня уже не применимо. ТАМ имеют тенденцию поляризоваться к М2 в ТМЭ, а их поверхностные рецепторы и секретируемые цитокины сходны с М2-подобными макрофагами.Между тем, ТАМ также экспрессируют некоторые M1-подобные маркеры макрофагов. Например, было обнаружено, что ТАМ iNOS (+) более благоприятны для прогноза онкологических больных [59]. Кроме того, эффект ТАМ в стимуляции активности Т-клеток с высокой экспрессией М2-подобных маркеров при асците яичников был сходен с эффектом М1-подобных ТАМ [60]. Следовательно, мы не можем просто классифицировать ТАМ как М1- или М2-подобные макрофаги и признать их функцию исключительно на основе экспрессии поверхностных маркеров.
Кроме того, эффект ТАМ в стимуляции активности Т-клеток с высокой экспрессией М2-подобных маркеров при асците яичников был сходен с эффектом М1-подобных ТАМ [60]. Следовательно, мы не можем просто классифицировать ТАМ как М1- или М2-подобные макрофаги и признать их функцию исключительно на основе экспрессии поверхностных маркеров.
Одним словом, биомаркеры ТАМ помогают выявить наличие и функцию макрофагов в опухолевых тканях (табл. 2), что может облегчить идентификацию, диагностику и лечение заболеваний.Поиск и открытие ценных биомаркеров ТАМ стали важным направлением исследований.
Взаимодействие ТАМ с другими иммунными и стромальными клетками в ТМЭ
ТАМ широко взаимодействуют с другими клетками микроокружения в ТМЭ (рис. 2). Ассоциированные с раком фибробласты (CAF), которые в основном происходят из нормальных фибробластов в интерстициальной ткани вокруг опухоли, тесно связаны с ТАМ [61]. Было обнаружено, что макрофаги, совместно культивируемые с CAF, повышали способность дифференцироваться в M2-подобные макрофаги посредством IL-6 и CSF-1, секретируемых CAF [62]. В эксперименте трехстороннего перекрестного общения между раковыми клетками, CAF и моноцитами группа с кондиционированной средой для раковых клеток (CM) показала самый высокий уровень М2-подобной дифференцировки [62]. В то же время CAF и TAM активируют друг друга, которые играют синергетическую роль в продвижении инвазии опухолевых клеток и ангиогенеза [63].
В эксперименте трехстороннего перекрестного общения между раковыми клетками, CAF и моноцитами группа с кондиционированной средой для раковых клеток (CM) показала самый высокий уровень М2-подобной дифференцировки [62]. В то же время CAF и TAM активируют друг друга, которые играют синергетическую роль в продвижении инвазии опухолевых клеток и ангиогенеза [63].
Основные роли ТАМ в опухолях. ТАМ состоят из макрофагов M1 и M2, причем M2 является доминирующим типом, индуцируемым компонентами TME. ТАМ способствуют пролиферации опухолевых клеток, ангиогенезу, метастазированию и иммуносупрессии посредством секреции цитокинов, высвобождения экзосом и ремоделирования ВКМ.В то же время опухолевые клетки могут также рекрутировать макрофаги и повторно обучать ТАМ
Для эндотелиальных клеток экзосомы, полученные из макрофагов, могут способствовать их пролиферации и увеличивать сосудистую плотность опухоли [64]. С другой стороны, исследования показали, что М2-подобные макрофаги при раке яичников напрямую контактируют с эндотелиальными клетками, чтобы усилить сосудистый барьер и снизить проницаемость сосудов, чтобы ограничить образование асцита [65].
Для Т-клеток ТАМ не только усиливали экспрессию PD-L1, но также снижали долю активации и пролиферации CD8 + Т-клеток [66].Некоторые исследования показали элиминацию Т-клеток при метастазах в печень, опосредованную макрофагами [67]. Регуляторные Т-клетки (Treg) и Т-хелперы 17 (Th27) представляют собой два подмножества Т-клеток CD4 + . Дисбаланс клеток Treg/Th27 является одной из характеристик опухолей [68]. При раке яичников экзосомы из макрофагов М2, которые содержат hsa-miR-21-5p, повышают соотношение Treg/Th27 и способствуют прогрессированию опухоли и метастазированию в брюшину [68]. С другой стороны, Treg-клетки могут преимущественно способствовать М2-подобным ТАМ, ингибируя секрецию IFN-γ CD8 + Т-клетками [69].Кроме того, активация естественных Т-клеток-киллеров увеличивает iNOS + CD206 — макрофагов M1 и контролирует рост солидных опухолей [70].
Супрессорные клетки миелоидного происхождения (MDSC) представляют собой незрелые клетки костного мозга с морфологическими и фенотипическими характеристиками нейтрофилов и моноцитов [71]. Блокирование ТАМ может способствовать компенсаторной инфильтрации G-MDSC, тем самым противодействуя снижению опухолевой нагрузки за счет ингибирования ТАМ [72]. Кроме того, экзосомы, высвобождаемые индуцированными опухолью мезенхимальными стволовыми клетками (MSC), ускоряли прогрессирование рака молочной железы, индуцируя M-MDSC для дифференцировки в макрофаги M2 при TME [73].Более того, данные также свидетельствуют о том, что опухолевые экзосомы также могут дифференцировать MDSC в макрофаги M2 [74].
Блокирование ТАМ может способствовать компенсаторной инфильтрации G-MDSC, тем самым противодействуя снижению опухолевой нагрузки за счет ингибирования ТАМ [72]. Кроме того, экзосомы, высвобождаемые индуцированными опухолью мезенхимальными стволовыми клетками (MSC), ускоряли прогрессирование рака молочной железы, индуцируя M-MDSC для дифференцировки в макрофаги M2 при TME [73].Более того, данные также свидетельствуют о том, что опухолевые экзосомы также могут дифференцировать MDSC в макрофаги M2 [74].
Связь ТАМ с гинекологическим раком и раком молочной железы
Новые данные свидетельствуют о том, что ТАМ участвуют в различных стадиях и аспектах развития опухоли, секретируя различные факторы. Эти молекулы, такие как IL-1/6/8/10, IGF-I, VEGF, EGF, MMP-7/9, uPA, TNF-α и TGF-β, в некоторой степени связаны с опухолями (рис. 3). , табл. 3) [75,76,77]. Здесь мы пытаемся определить влияние ТАМ на прогрессирование опухоли с помощью следующих аспектов (таблица 3).
Рис. 3
3 Взаимодействие ТАМ и других иммунных и стромальных клеток при ТМЭ
Таблица 3 Роль производных ТАМ цитокинов при гинекологическом раке и раке молочной железы контролировать и размножаться бесконечно, что затрудняет их удаление и разрушает окружающие нормальные ткани. Различные исследования показали, что ТАМ секретируют IL-6 [78, 79], который является цитокином, принадлежащим к семейству интерлейкинов, связанных с онкогенезом.Доказано, что IL-6 играет жизненно важную роль посредством передачи сигнала IL-6-JAK-STAT [80,81,82], а активация STAT3 является важным фактором, влияющим на пролиферацию, дифференцировку и выживание клеток [83]. Уровень белка STAT3 и состояние фосфорилирования были значительно индуцированы в ВПЧ-положительных клетках рака шейки матки по сравнению с ВПЧ-отрицательными, что, как было показано, опосредовано онкобелком Е6 ВПЧ [84]. Между тем, Луан и др. наблюдали синхронное увеличение экспрессии IL-6R в клетках рака шейки матки, что, по-видимому, неблагоприятно влияет на прогноз пациентов [85]. Это говорит о том, что IL-6, секретируемый ТАМ, может влиять на пролиферацию, миграцию и адгезию опухоли посредством активации IL-6R. Однако активация пути IL6/STAT3, которая была обнаружена в совершенно новом исследовании, способствовала метастазированию внутрипротокового рака молочной железы ER + in vivo и, поразительно, представляла собой еще один онкогенный путь, который был относительно независимым от пути ER [86].
Это говорит о том, что IL-6, секретируемый ТАМ, может влиять на пролиферацию, миграцию и адгезию опухоли посредством активации IL-6R. Однако активация пути IL6/STAT3, которая была обнаружена в совершенно новом исследовании, способствовала метастазированию внутрипротокового рака молочной железы ER + in vivo и, поразительно, представляла собой еще один онкогенный путь, который был относительно независимым от пути ER [86]. Другой член семейства интерлейкинов, CXCL8 (известный также как IL-8), активно участвует в онкогенезе из-за его плюрипотентности, включая ангиогенез и митогенные эффекты [87].CXCL8 в основном секретируется мононуклеарными макрофагами. Сообщалось, что ТАМ подавляют экспрессию ERα в тканях карциномы эндометрия за счет избыточной секреции CXCL8 через гомеодоменовый фактор транскрипции HOXB13, что связано с инвазией опухоли и плохим прогнозом [88]. Параллельные результаты были обнаружены при раке молочной железы ERα + . Повышающая регуляция HOXB13 ускоряет пролиферацию клеток и метастазирование как in vitro, так и in vivo за счет прямого подавления ERα, которое можно ингибировать путем снижения экспрессии mTOR [89]. Между тем, одно исследование показало, что экспрессия ERα при раке шейки матки снижается более чем в 15 раз по мере того, как рак шейки матки прогрессирует от нормального до рака [90]. Однако о взаимосвязи между снижением ERα и ТАМ в клетках рака шейки матки не сообщалось.
Между тем, одно исследование показало, что экспрессия ERα при раке шейки матки снижается более чем в 15 раз по мере того, как рак шейки матки прогрессирует от нормального до рака [90]. Однако о взаимосвязи между снижением ERα и ТАМ в клетках рака шейки матки не сообщалось.
Кроме того, было продемонстрировано, что эстроген, IL-6 и CXCL8 способны взаимодействовать друг с другом, регулируя рост эпителиального рака яичников посредством эффекта каскадной амплификации через путь ERα [91, 92].
Нарушение регуляции ангиогенеза и лимфангиогенеза
Увеличение деления и роста раковых клеток указывает на то, что им требуется больше питательных веществ из крови. Следовательно, опухолевой ангиогенез играет ключевую роль в росте, инвазии и метастазировании солидных опухолей. Недавно было показано, что ТАМ тесно связаны с ангиогенезом и лимфангиогенезом посредством секреции VEGF-A, VEGF-C, VEGF-D, EGF, плацентарного фактора роста (PlGF), TGF-β, TNF-α, IL-1β, IL-8, CCL2, CXCL8 и CXCL12 [93]. При раке молочной железы инфильтрация ТАМ тесно связана с экспрессией VEGF и плотностью микрососудов (MVD), что совместно влияет на прогноз пациентов [94]. Кроме того, предыдущие исследования подтвердили, что VEGF-C, VEGF-D и их рецептор VEGFR-3 положительно коррелируют с различными стадиями поражения шейки матки, что способствует развитию лимфангиона [95, 96]. Интересно, что экспрессия VEGF-C и VEGF-D значительно увеличивалась после прогрессирования заболевания до стадии CIN3 [96].Однако при раке эндометрия был сделан противоположный вывод [95]. В одном исследовании сообщалось, что более низкий уровень VEGF-C был обнаружен в образцах рака эндометрия [95]. Мы подозреваем, что противоречия могут объясняться характеристиками различных опухолей и ограниченным размером выборки.
При раке молочной железы инфильтрация ТАМ тесно связана с экспрессией VEGF и плотностью микрососудов (MVD), что совместно влияет на прогноз пациентов [94]. Кроме того, предыдущие исследования подтвердили, что VEGF-C, VEGF-D и их рецептор VEGFR-3 положительно коррелируют с различными стадиями поражения шейки матки, что способствует развитию лимфангиона [95, 96]. Интересно, что экспрессия VEGF-C и VEGF-D значительно увеличивалась после прогрессирования заболевания до стадии CIN3 [96].Однако при раке эндометрия был сделан противоположный вывод [95]. В одном исследовании сообщалось, что более низкий уровень VEGF-C был обнаружен в образцах рака эндометрия [95]. Мы подозреваем, что противоречия могут объясняться характеристиками различных опухолей и ограниченным размером выборки.
С другой стороны, ТАМ экспрессировали HIF-1α для активного ангиогенеза [97]. Было доказано, что при раке молочной железы HIF-1α-стабилизирующая длинная некодирующая РНК (HISLA) предотвращает инактивацию HIF-1α от гидроксилирования пролилгидроксилазным доменом 2 (PHD2) [98]. Более того, HISLA, продуцируемый внеклеточными везикулами, высвобождаемыми ТАМ, может ингибировать взаимодействие между HIF-1α и PHD2 и способствовать стабильности HIF-1α в TME [97].
Более того, HISLA, продуцируемый внеклеточными везикулами, высвобождаемыми ТАМ, может ингибировать взаимодействие между HIF-1α и PHD2 и способствовать стабильности HIF-1α в TME [97].
В исследовании трансцеломического метастазирования рака яичников ТАМ в опухолевых скоплениях (сфероидах) мотивировали экспрессию EGF на ранней стадии метастазирования, которая была интегрирована с активированным EGFR опухоли, дополнительно поддерживая пролиферацию и миграцию опухолевых клеток за счет усиления передачи сигналов VEGF/VEGFR в окружающих опухолевых клетках [99].Также наблюдалось, что EGF увеличивает интегрин αMβ2 на ТАМ и ICAM-1 на опухолевых клетках, способствуя адгезии между ТАМ и опухолевыми клетками [99]. Исихара и др. обнаружили, что белок синдрома Вискотта-Олдрича (WASp) позволяет ТАМ секретировать EGF и хемотаксически приближаться к экспрессирующим CSF-1 клеткам рака молочной железы, чтобы регулировать метастазирование опухоли [100].
Содействие ЕМТ и метастазированию опухоли
ЕМТ и ремоделирование внеклеточного матрикса представляют собой важные звенья в стимулировании метастазирования опухоли. Цзин и др. показали, что под действием агонистов ERα макрофаги ERα + M2 значительно активировали CCL18 и активировали сигнальный путь mTOR/KIF5B в раковых клетках эндометрия, способствуя ЭМП [101].
Цзин и др. показали, что под действием агонистов ERα макрофаги ERα + M2 значительно активировали CCL18 и активировали сигнальный путь mTOR/KIF5B в раковых клетках эндометрия, способствуя ЭМП [101].
Матриксные металлопротеиназы (ММП), которые широко представлены в ТМЭ, могут не только способствовать ЭМП, но и способствовать лимфангиогенезу совместно с VEGF [93]. Недавно одно исследование показало, что М2-подобные макрофаги секретируют большое количество MMP-9, что способствует прогрессированию опухоли как при раке молочной железы, так и при раке толстой кишки, что считается прогностическими маркерами и терапевтическими мишенями [102, 103].При раке молочной железы ER + MMP-9, секретируемый ТАМ, был связан с худшей общей выживаемостью, но не при ER-отрицательном раке молочной железы или трижды отрицательном раке молочной железы (TNBC) [104]. Вен и др. сообщили, что повышение уровня MMP-7 в макрофагах, которые совместно культивировали с клетками рака яичников, происходило через путь p38 и способствовало инфильтрации макрофагов с образованием TME [105]. В то же время большинство исследований подтверждают мнение о том, что повышенный уровень ММР-9 также может способствовать развитию рака шейки матки [106, 107], хотя в нескольких статьях высказывается предположение, что ММР-9 помогает прогнозировать рак шейки матки [103]. ].Поэтому, принимая во внимание различия в исследованиях, влияние ММП на рак шейки матки нуждается в дальнейшей оптимизации.
В то же время большинство исследований подтверждают мнение о том, что повышенный уровень ММР-9 также может способствовать развитию рака шейки матки [106, 107], хотя в нескольких статьях высказывается предположение, что ММР-9 помогает прогнозировать рак шейки матки [103]. ].Поэтому, принимая во внимание различия в исследованиях, влияние ММП на рак шейки матки нуждается в дальнейшей оптимизации.
Кроме того, ТАМ также играют огромную роль в стимулировании метастазирования опухолевых клеток, высвобождая активатор плазминогена урокиназного типа (uPA) и усиливая экспрессию uPA-рецептора (uPAR), который относится к системе фибринолиза [108]. Комбинация uPA и uPAR инициирует реакции изменения формы внеклеточного матрикса путем деградации соответствующих компонентов и впоследствии приводит к отдаленной миграции опухолевых клеток из первичного очага [109].Кроме того, повышенный уровень uPAR также может вызывать агрегацию макрофагов в опухолевой ткани, формируя положительную обратную связь [110]. При карциноме протоков молочной железы in situ и инвазивных карциномах молочной железы моноциты крови с более высокими уровнями uPAR могут избирательно рекрутироваться в раковые ткани, что также вызывает повышенные уровни uPAR в ТАМ посредством паракринного действия [110].
При карциноме протоков молочной железы in situ и инвазивных карциномах молочной железы моноциты крови с более высокими уровнями uPAR могут избирательно рекрутироваться в раковые ткани, что также вызывает повышенные уровни uPAR в ТАМ посредством паракринного действия [110].
Как член трансформирующих факторов роста, TGF-β является многофункциональным цитокином. Он действует как мощный супрессор опухоли на ранней стадии рака, в то время как позже эти цитостатические эффекты становятся устойчивыми, и TGF-β участвует в индукции ЭМП [111].Что касается серозного рака яичников высокой степени, то асцит с высоким уровнем ТАМ и TGF-β1 способствует миграции раковых клеток [112]. При раке шейки матки GDF15, который принадлежит к суперсемейству TGF-β, фосфорилирует сигнальные пути PI3K/AKT и MAPK/ERK через ErbB2, члена семейства EGFR, для достижения цели, способствующей прогрессированию опухоли [113].
Формирование иммуносупрессии
Опухолевые клетки избегают распознавания и нападения со стороны иммунной системы организма с помощью множества механизмов. IL-10 представляет собой многоклеточный цитокин с иммунодепрессивным действием и может ингибировать доставку антигена системой мононуклеарных фагоцитов [114, 115]. Одно предыдущее исследование показало, что IL-10 способствует трансформации макрофагов в M2-подобные макрофаги при TME, а поляризованные M2-подобные макрофаги, в свою очередь, высоко секретируют IL-10 [116]. Сообщалось, что при поражениях шейки матки количество IL-10 в ТМЭ ассоциировалось с типом инфекции ВПЧ и степенью прогрессирования поражений в рак шейки матки.Более того, при значительном увеличении уровня IL-10 было обнаружено усиление экспрессии HPV E6 и E7 [117]. Кроме того, доказана роль IL-10 в противоопухолевых Т-клетках [119, 120]. При раке молочной железы сверхэкспрессия IL-10, происходящая из макрофагов, приводила к развитию опухоли из-за неэффективности CD8 + Т-клеточно-зависимых ответов на химиотерапию [121]. Как Чу и др. сообщалось, что IL-10 также был независимым фактором неблагоприятного прогноза у пациентов с ER-отрицательным раком молочной железы [122].
IL-10 представляет собой многоклеточный цитокин с иммунодепрессивным действием и может ингибировать доставку антигена системой мононуклеарных фагоцитов [114, 115]. Одно предыдущее исследование показало, что IL-10 способствует трансформации макрофагов в M2-подобные макрофаги при TME, а поляризованные M2-подобные макрофаги, в свою очередь, высоко секретируют IL-10 [116]. Сообщалось, что при поражениях шейки матки количество IL-10 в ТМЭ ассоциировалось с типом инфекции ВПЧ и степенью прогрессирования поражений в рак шейки матки.Более того, при значительном увеличении уровня IL-10 было обнаружено усиление экспрессии HPV E6 и E7 [117]. Кроме того, доказана роль IL-10 в противоопухолевых Т-клетках [119, 120]. При раке молочной железы сверхэкспрессия IL-10, происходящая из макрофагов, приводила к развитию опухоли из-за неэффективности CD8 + Т-клеточно-зависимых ответов на химиотерапию [121]. Как Чу и др. сообщалось, что IL-10 также был независимым фактором неблагоприятного прогноза у пациентов с ER-отрицательным раком молочной железы [122].
В тканях рака яичников, Qu et al. обнаружили повышенную экспрессию PD-L1 на поверхности ТАМ, которые стимулировали апоптоз Т-клеток для подавления иммунитета [123]. Современные исследования показали, что избыточная экспрессия белка 4 придатка яичка человека (HE4) в клетках рака яичников может быть одной из причин повышения уровня PD-L1 макрофагами [124]. В то же время ТАМ, а также IFN-γ, TNF-α, IL-10 и особенно IL-6, секретируемые ТАМ, индуцировали экспрессию PD-L1 как в клеточной мембране, так и в цитоплазме клеток рака яичников, что ингибировало функцию CD8 + Т-клеток и способствовали росту опухоли [125].Такой же ингибирующий эффект на Т-клетки был также обнаружен при раке шейки матки, ассоциированном с ВПЧ16, через макрофаги М2 [119]. В последнее время в исследованиях прогрессирования стромальных опухолей ингибирование Т-клеток CD8 + PD-1/PD-L1 может осуществляться через сигнальный путь PI3K/Akt/mTOR [126]. Кроме того, гипоксические макрофаги способствуют высвобождению экзосом, содержащих миР-223. Он может быть перенесен в совместно культивируемые эпителиальные раковые клетки яичников для повышения лекарственной устойчивости посредством пути PTENPI3K/AKT как in vivo, так и in vitro [127].Как упоминалось выше, сопровождающаяся гипоксией TME, VEGF сверхэкспрессируется в макрофагах. Он не только управляет ангиогенезом, но и подавляет иммунные клетки, которые не могут выполнять свою обычную роль [128]. Для Т-клеток VEGF блокирует дифференцировку Т-клеток-предшественников в CD4 + и CD8 + лимфоциты, ослабляет иммунную функцию Т-клеток. С другой стороны, VEGF увеличивает проникновение ТАМ в участки опухоли, что формирует положительную обратную связь от VEGF к ТАМ [129].
Он может быть перенесен в совместно культивируемые эпителиальные раковые клетки яичников для повышения лекарственной устойчивости посредством пути PTENPI3K/AKT как in vivo, так и in vitro [127].Как упоминалось выше, сопровождающаяся гипоксией TME, VEGF сверхэкспрессируется в макрофагах. Он не только управляет ангиогенезом, но и подавляет иммунные клетки, которые не могут выполнять свою обычную роль [128]. Для Т-клеток VEGF блокирует дифференцировку Т-клеток-предшественников в CD4 + и CD8 + лимфоциты, ослабляет иммунную функцию Т-клеток. С другой стороны, VEGF увеличивает проникновение ТАМ в участки опухоли, что формирует положительную обратную связь от VEGF к ТАМ [129].
В совокупности ТАМ участвуют в секреции цитокинов и факторов роста, влияя на рост, метастазирование и прогрессирование опухоли.Различные типы гинекологического рака и рака молочной железы имеют сходные или специфические характеристики ТАМ (рис. 4). Необходимо провести всесторонние исследования, чтобы найти больше факторов, секретируемых ТАМ, которые принимают участие в регуляции опухоли, что позволит лучше понять целевые методы лечения ТАМ.
Характеристика ТАМ при гинекологическом раке и раке молочной железы. a ER + пациентов с раком молочной железы имели более низкие показатели выживаемости. ТАМ усиливали экспрессию HOXB13 и подавляли экспрессию IL-6 и ER. b Каскад эстрогена, CXCL8 и IL-6 регулирует рост клеток рака яичников. c ТАМ стимулировали ЭМП при карциноме эндометрия через HOXB13. d Тип инфекции ВПЧ ассоциировался с количеством ТАМ и экспрессией IL-10. ТАМ повышали экспрессию IL-6, MMP-9 и GDF15 при раке шейки матки
Терапия, направленная на ТАМ, при гинекологическом раке и раке молочной железы
имеют многочисленные функции в пролиферации рака, метастазировании и иммунной резистентности.В настоящее время проведены различные клинические испытания ТАМ-таргетной терапии при гинекологическом раке и раке молочной железы (https://clinicaltrials.gov/, табл. 4). После приведенного выше обсуждения роли ТАМ мы глубоко осознаем, что вмешательство ТАМ, включая истощение макрофагов, ингибирование про-ТАМ или активацию анти-ТАМ, может быть многообещающим терапевтическим подходом (рис. 5, таблица 5).
5, таблица 5).
Возможные целевые терапевтические стратегии для ТАМ. a Прямое блокирование хемокинов макрофагов или ингибирование их рецепторов. b Использование клодроната для уничтожения макрофагов. c Увеличение количества M1 или продвижение преобразования M2 в M1 для повышения соотношения M1/M2. d Блокирование секреции ТАМ, действующих на раковые клетки. e Терапия макрофагами CAR. f Иммунная вакцина на основе ТАМ s. г ТАМ нанобиотехнология
Таблица 5 Стратегии таргетной терапии ТАМУменьшение образования ТАМ
Поляризация ТАМ регулируется различными цитокинами микроокружения, факторами роста и сигналами как опухолевых, так и стромальных клеток, что представляет собой процесс взаимной конверсии между M1 и M2 [14]. Учитывая обширную роль ТАМ в ТМЭ, контроль количества ТАМ, другими словами, подавление поляризации макрофагов М2 или направление макрофагов к дифференцировке в М1-подобные представляют большой интерес в качестве новой стратегии лечения.
Учитывая обширную роль ТАМ в ТМЭ, контроль количества ТАМ, другими словами, подавление поляризации макрофагов М2 или направление макрофагов к дифференцировке в М1-подобные представляют большой интерес в качестве новой стратегии лечения.
С точки зрения снижения количества ТАМ клодронат стал основным средством выбора для истощения макрофагов [130]. При раке шейки матки Lepique et al. вводили клодронатсодержащие липосомы (CL) в мышиной модели для потребления макрофагов путем накопления метиленсодержащих аналогов АТФ в цитоплазме.Хотя КЛ не регулирует напрямую цитоактив раковых клеток, сниженное количество макрофагов ингибирует рост опухоли in vivo и способствует инфильтрации опухолей ВПЧ16 Е7(49-57)-специфическими CD8-лимфоцитами [119]. Тем не менее, терапия все еще нуждается в дальнейшей оптимизации для удовлетворения клинических потребностей, например, как сосредоточиться на ТАМ, не мешая другим макрофагам в нормальных тканях организма.
С другой стороны, препятствование рекрутированию ТАМ в опухолевой ткани имеет широкие перспективы применения в будущем за счет предотвращения рекрутирования ТАМ с помощью цитокинов. Энгелетин (ЭНГ, дигидрокемпферол 3-рамнозид) представляет собой вещество, обладающее противовоспалительными свойствами [131, 132]. При раке шейки матки одно исследование показало, что ENG значительно снижает секрецию CCL2 за счет блокирования сигнального пути NF-κB, что можно рассматривать как ингибитор NF-κB [133]. Благодаря ЭНГ моноциты периферической крови меньше рекрутировались на ТМЭ. Ингибиторы CSF1/CSF1R, такие как MCS110, пексидартиниб (PLX3397, PLX108-01) и эмактузумаб (RG7155), изучаются в клинических испытаниях при солидных опухолях, включая рак яичников и эндометрия [43, 134].Хойсинквельд и др. сообщили, что определенная концентрация PGE2 и IL-6 может вызвать дифференцировку макрофагов CD14 + в макрофаги CD163 + M2 [34]. К счастью, действие ПГЕ и ИЛ-6 полностью блокировалось ингибитором кокс-оксидазы и антителами к ИЛ-6 [34]. Точно так же использование ингибиторов различных профакторов, таких как IL-4, IL-10, TGF-β и IL-13, может в определенной степени уменьшить миграцию ТАМ в направлении микроокружения опухоли [135, 136, 137].
Энгелетин (ЭНГ, дигидрокемпферол 3-рамнозид) представляет собой вещество, обладающее противовоспалительными свойствами [131, 132]. При раке шейки матки одно исследование показало, что ENG значительно снижает секрецию CCL2 за счет блокирования сигнального пути NF-κB, что можно рассматривать как ингибитор NF-κB [133]. Благодаря ЭНГ моноциты периферической крови меньше рекрутировались на ТМЭ. Ингибиторы CSF1/CSF1R, такие как MCS110, пексидартиниб (PLX3397, PLX108-01) и эмактузумаб (RG7155), изучаются в клинических испытаниях при солидных опухолях, включая рак яичников и эндометрия [43, 134].Хойсинквельд и др. сообщили, что определенная концентрация PGE2 и IL-6 может вызвать дифференцировку макрофагов CD14 + в макрофаги CD163 + M2 [34]. К счастью, действие ПГЕ и ИЛ-6 полностью блокировалось ингибитором кокс-оксидазы и антителами к ИЛ-6 [34]. Точно так же использование ингибиторов различных профакторов, таких как IL-4, IL-10, TGF-β и IL-13, может в определенной степени уменьшить миграцию ТАМ в направлении микроокружения опухоли [135, 136, 137].
При раке шейки матки, по сравнению с профилактической вакциной против ВПЧ, которая уже повсеместно используется в качестве контрмеры против рака шейки матки, связанного с ВПЧ [138], терапевтическая вакцина против ВПЧ все еще находится в разработке.В испытании вакцины с участием пептида HPV16 E7 43–77 наблюдалось снижение количества макрофагов М2 и экспрессии цитокинов, связанных с макрофагами М2, таких как IL10, TGF-β, MMP-2, MMP-9 и VEGF. в группе вакцин [139], в то время как хемокины, связанные с макрофагами M1 (такие как CXCL-9 и CXCL-10), регулировались с повышением [139].
Реактивация фагоцитоза ТАМ M1
Как упоминалось выше, низкая частота M1/M2 часто предсказывает неблагоприятный прогноз для пациентов [140]. Следовательно, попытка увеличить количество M1 или улучшить соотношение M1/M2 является реальным способом возможного лечения [141].Индоламин-2,3-диоксигеназа (IDO1) является ключевым ферментом для деградации аминокислоты триптофана через кинурениновый путь [142], который может регулировать макрофаги и ингибировать иммунную функцию Т-клеток при различных заболеваниях и раке [142, 143, 144, 145]. При раке шейки матки существующие исследования показали, что IFN-γ активирует аутофагию клеток рака шейки матки и реактивацию фагоцитоза макрофагов за счет сверхэкспрессии IDO1 и метаболизма кинуренина [146, 147]. Помимо рака шейки матки, Feng et al.подтвердили, что высокая экспрессия IDO1 свидетельствует о лучшем прогнозе выживания при раке молочной железы и яичников [148]. Хотя эти исследования показали, что повышенная регуляция IDO1 в некоторой степени полезна для подавления рака, другие исследования показали, что сверхэкспрессия IDO1 связана с иммуносупрессией и играет неблагоприятную роль в процессе развития опухоли [149, 150].
При раке шейки матки существующие исследования показали, что IFN-γ активирует аутофагию клеток рака шейки матки и реактивацию фагоцитоза макрофагов за счет сверхэкспрессии IDO1 и метаболизма кинуренина [146, 147]. Помимо рака шейки матки, Feng et al.подтвердили, что высокая экспрессия IDO1 свидетельствует о лучшем прогнозе выживания при раке молочной железы и яичников [148]. Хотя эти исследования показали, что повышенная регуляция IDO1 в некоторой степени полезна для подавления рака, другие исследования показали, что сверхэкспрессия IDO1 связана с иммуносупрессией и играет неблагоприятную роль в процессе развития опухоли [149, 150].
Раковые клетки экспрессируют CD47 для защиты клеток от фагоцитоза путем связывания и активации рецептора SIRPα на макрофагах для ингибирования фагоцитоза макрофагов.Воздействие на ось CD47-SIRPα стало потенциальным терапевтическим механизмом в различных доклинических моделях путем стимуляции фагоцитоза раковых клеток in vitro и in vivo [151, 152]. Повышенная экспрессия CD47 была обнаружена почти во всех типах опухолевых клеток, что было связано с плохой выживаемостью и плохим прогнозом [153,154,155]. Хуанг и др. сконструировали новый онколитический аденовирус, несущий слитый ген сигнального регуляторного белка-α (SIRPα)-IgG1 Fc (названный SG635-SF), который значительно ингибировал рост клеток рака яичников in vivo [156].Саманта и др. сообщили, что обработка клеток TNBC карбоплатином, доксорубицином, гемцитабином или паклитакселом способствовала обогащению CD47 + CD73 + PDL1 + клеток TNBC и способствовала ускользанию опухоли от иммунного ответа, которое можно было устранить путем ингибирования HIF [157]. Основываясь на решающей роли оси CD47-SIRPα в ускользании опухоли от иммунного ответа, в настоящее время проводятся клинические испытания антител против CD47 с участием таких препаратов, как Hu5F9-G4, HX009 и AK117. Исследование рака молочной железы показало, что CD47 и HER2 активируют друг друга на уровне транскрипции [158].Двойное антитело, блокирующее CD47 и HER2, не только ингибировало образование опухолевых клонов, но и усиливало опосредованную макрофагами атаку [158].
Хуанг и др. сконструировали новый онколитический аденовирус, несущий слитый ген сигнального регуляторного белка-α (SIRPα)-IgG1 Fc (названный SG635-SF), который значительно ингибировал рост клеток рака яичников in vivo [156].Саманта и др. сообщили, что обработка клеток TNBC карбоплатином, доксорубицином, гемцитабином или паклитакселом способствовала обогащению CD47 + CD73 + PDL1 + клеток TNBC и способствовала ускользанию опухоли от иммунного ответа, которое можно было устранить путем ингибирования HIF [157]. Основываясь на решающей роли оси CD47-SIRPα в ускользании опухоли от иммунного ответа, в настоящее время проводятся клинические испытания антител против CD47 с участием таких препаратов, как Hu5F9-G4, HX009 и AK117. Исследование рака молочной железы показало, что CD47 и HER2 активируют друг друга на уровне транскрипции [158].Двойное антитело, блокирующее CD47 и HER2, не только ингибировало образование опухолевых клонов, но и усиливало опосредованную макрофагами атаку [158]. При TNBC антитело к CD47 ингибировало дифференцировку раковых стволовых клеток (CSC) и подавляло экспрессию EGFR [159]. Кроме того, иммунолипосома (ILips) в сочетании с анти-CD47 и паклитакселом также оказывает синергетическое противоопухолевое действие на превращение М2 в М1 и усиление системного Т-клеточного иммунного ответа [160].
При TNBC антитело к CD47 ингибировало дифференцировку раковых стволовых клеток (CSC) и подавляло экспрессию EGFR [159]. Кроме того, иммунолипосома (ILips) в сочетании с анти-CD47 и паклитакселом также оказывает синергетическое противоопухолевое действие на превращение М2 в М1 и усиление системного Т-клеточного иммунного ответа [160].
High-mobility group box 1 (HMGB1) представляет собой негистоновый хроматин-связывающий белок, широко распространенный в ядре и цитоплазме, который, как было доказано, активирует Treg-клетки и макрофаги M2 в базальноподобных клетках рака молочной железы [ 161].Более того, этот эффект может быть заблокирован ингибиторами HMGB1 A Box, RAP или EP, что приводит к уменьшению доли Treg-клеток и увеличению соотношения M1/M2 [161]. Аналогичным образом, в исследовании традиционной китайской медицины (ТКМ) лечения рака яичников было обнаружено, что астрагалозид IV ослабляет поляризацию макрофагов М2 путем ингибирования передачи сигнала HMGB1-TLR4 [162].
Toll-подобные рецепторы (TLR) относятся к консервативным мембранным белкам, которые распознают патогены [163]. Исследование подтвердило, что TLR7, связанный с ингибиторами TGF-β рецептора I SB431542 (SB4x), совместно провоцирует поляризацию клеток M1 и CD4 + , CD8 + и CD19 + [164].Трэверс и др. обнаружили, что DNMTi 5-азацитидин (5AZA-C) активирует передачу сигналов интерферона I типа при раке яичников, катализирует количество IFN-γ + иммунных клеток и снижает процент макрофагов в TME [165]. Кроме того, 5AZA-C в сочетании с ингибитором орнитиндекарбоксилазы α-дифторметилхлором (DFMO) значительно уменьшал количество макрофагов M2 и увеличивал количество макрофагов M1 [165]. Чжао и др. обнаружили, что колоректальный рак, обработанный цетуксимабом, реполяризует ТАМ с макрофагов M2 на M1 зависимым от IL6 образом [166].
Исследование подтвердило, что TLR7, связанный с ингибиторами TGF-β рецептора I SB431542 (SB4x), совместно провоцирует поляризацию клеток M1 и CD4 + , CD8 + и CD19 + [164].Трэверс и др. обнаружили, что DNMTi 5-азацитидин (5AZA-C) активирует передачу сигналов интерферона I типа при раке яичников, катализирует количество IFN-γ + иммунных клеток и снижает процент макрофагов в TME [165]. Кроме того, 5AZA-C в сочетании с ингибитором орнитиндекарбоксилазы α-дифторметилхлором (DFMO) значительно уменьшал количество макрофагов M2 и увеличивал количество макрофагов M1 [165]. Чжао и др. обнаружили, что колоректальный рак, обработанный цетуксимабом, реполяризует ТАМ с макрофагов M2 на M1 зависимым от IL6 образом [166].
В приведенном выше описании биомаркеров ТАМ мы упоминали, что стимуляция макрофагов различными факторами может влиять на экспрессию рецепторов (Tyro3, Axl и MerTK рецептор) на поверхности макрофагов. Сан и др. доказали, что экспрессия Axl и его лигандов Gas6 была аномальной при высокодифференцированном раке эндометрия, что может быть связано с опухолевой прогрессией и ингибированием апоптоза [168]. Однако Goyette et al. обнаружили, что высокие уровни белка AxL были связаны с метастазами опухоли при раке молочной железы HER2 + , который не зависел от Gas6 [169].Принимая во внимание, что рецепторы ТАМ опосредуют рост, инвазию и метастазирование опухоли, исследования ингибиторов рецепторов бурно развиваются [167]. Несколько исследований показали, что ингибитор киназы pan TAMs BMS-777607 может ингибировать активацию рецептора TAMs, дополнительно блокировать сигнальный путь PD-L1 и подавлять прогрессирование опухоли [170], в то время как селективный ингибитор Axl BGB324, по-видимому, индуцирует апоптоз, ингибируя путь PI3K [170]. 171]. Кроме того, функция AVB-S6-500 была подтверждена в клинических испытаниях в качестве слитого белка против Axl Fc при эпителиальном раке яичников, первичном раке брюшины и раке фаллопиевых труб [172].
Однако Goyette et al. обнаружили, что высокие уровни белка AxL были связаны с метастазами опухоли при раке молочной железы HER2 + , который не зависел от Gas6 [169].Принимая во внимание, что рецепторы ТАМ опосредуют рост, инвазию и метастазирование опухоли, исследования ингибиторов рецепторов бурно развиваются [167]. Несколько исследований показали, что ингибитор киназы pan TAMs BMS-777607 может ингибировать активацию рецептора TAMs, дополнительно блокировать сигнальный путь PD-L1 и подавлять прогрессирование опухоли [170], в то время как селективный ингибитор Axl BGB324, по-видимому, индуцирует апоптоз, ингибируя путь PI3K [170]. 171]. Кроме того, функция AVB-S6-500 была подтверждена в клинических испытаниях в качестве слитого белка против Axl Fc при эпителиальном раке яичников, первичном раке брюшины и раке фаллопиевых труб [172].
Кроме того, Zhao et al. продемонстрировали, что стимулирование экспрессии длинной некодирующей РНК (днРНК)-Xist в макрофагах M1 и подавление экспрессии микроРНК (miR)-101 в макрофагах M2 может способствовать конверсии макрофагов типа M2 в M1, что может играть полезную роль в ингибирование пролиферации и миграции рака молочной железы и яичников [173].
Ингибирование цитокинов макрофагов
Как указывалось ранее, макрофаги способствуют росту и метастазированию опухолевых клеток, секретируя цитокины и факторы роста.Следовательно, блокирование соответствующей передачи сигнала для вмешательства в цитокины является еще одним методом ограничения роли ТАМ.
Недавние исследования показали, что при раке шейки матки Poly(I:C), адъювант противораковой вакцины, может действовать через сигнальный путь NF-kB, способствуя экспрессии цитокинов M1-типа (таких как IL-1β и IL -6) и подавляют экспрессию цитокинов М2-типа (таких как IL-10 и CCL22) [174]. Другое исследование показало, что ТАМ аутокринно VEGF также действует через путь NF-κB [175].Блокирование передачи ТАМ по внутреннему пути NF-κB также может ослабить проангиогенезный эффект VEGF. Кроме того, в макрофагах с нокаутом MMP-12 были снижены экспрессия и фосфорилирование p38 и ERK1/2, а также снижена секреция IL-1β, IL-6, TNF-α, CXCL1 и CXCL3 [176]. В недавних исследованиях сообщалось о трех новых селективных ингибиторах гидроксиэфира ММР-12 (CGA, CGA-1 и AGA) и двух радиоактивных индикаторах 99mTc, полученных из AGA [99mTc]-AGA-1 и [99mTc]-AGA-2 [177]. ].Кроме того, ингибитор ММП широкого спектра действия СР-471/474 и двойной ингибитор ММП-9/ММП-12 (AZ11557272) показали хорошее ингибирование ММП [178, 179]. Однако необходимы дальнейшие клинические испытания селективных ингибиторов ММР для снижения токсичности лекарств. Что касается uPA, его ингибитор, 4-йодобензо[b]тиофен-2-карбоксамидин (B-428), как было показано, препятствует росту и метастазированию рака молочной железы в комбинации с тамоксифеном еще с 1990-х годов, что превосходит их разделительные эффекты [180].
].Кроме того, ингибитор ММП широкого спектра действия СР-471/474 и двойной ингибитор ММП-9/ММП-12 (AZ11557272) показали хорошее ингибирование ММП [178, 179]. Однако необходимы дальнейшие клинические испытания селективных ингибиторов ММР для снижения токсичности лекарств. Что касается uPA, его ингибитор, 4-йодобензо[b]тиофен-2-карбоксамидин (B-428), как было показано, препятствует росту и метастазированию рака молочной железы в комбинации с тамоксифеном еще с 1990-х годов, что превосходит их разделительные эффекты [180].
Основываясь на важной роли ИЛ-6, полученного из ТАМ, при раке, терапевтические стратегии, нацеленные на метаболический путь ИЛ-6, находятся в активной разработке.Силтуксимаб относится к моноклональным антителам (МАТ), блокирующим рецептор ИЛ-6 [181]. Было обнаружено, что при лечении силтуксимабом рака яичников и ERα-положительного рака молочной железы раковые клетки снижают продукцию IL-6, что ингибирует рост опухоли, инвазию ТАМ и ангиогенез [182, 183, 184]. Аналогичным образом тоцилизумаб был эффективен при раке яичников, раке шейки матки, раке эндометрия и раке молочной железы [185, 186, 187, 188]. Кроме того, в одном исследовании было обнаружено, что цетуксимаб может также усиливать противоопухолевую функцию макрофагов через путь IL-6 [166].
Кроме того, в одном исследовании было обнаружено, что цетуксимаб может также усиливать противоопухолевую функцию макрофагов через путь IL-6 [166].
Для борьбы с повышенной экспрессией TGF-β исследователи использовали антитела для блокирования как рецептора, так и лиганда, такие как фрезолимумаб (GC1008) и LY3022859 [189]. Также полезно изменить структуру или состав лиганда или рецептора, чтобы повлиять на их связывание [189]. Агенты, нацеленные на TGF-β, такие как LY2109761 и 1D11, доказали свою эффективность в ингибировании метастазирования при метастатическом раке молочной железы [190].
ИЛ-10 представляет собой привлекательную терапевтическую мишень из-за его функции иммуносупрессии и участия в онкогенезе.Один из предыдущих экспериментов показал, что mAb, блокирующие рецептор IL-10 (αIL-10R; клон 1B1.3A), усиливали терапевтический эффект химиотерапии на модели рака молочной железы в зависимости от CD8 + Т-клеток [121]. Кроме того, Алотаиби и соавт. использовали РНКи для снижения секреции IL-10, что приводило к ускорению апоптоза клеток рака молочной железы с подавлением пути PI3K/Akt [136].
Таким образом, терапия, нацеленная на цитокины и факторы роста, может проводиться следующими способами.Во-первых, на генном уровне технология РНК-интерференции может влиять на экспрессию молекул. Кроме того, экспрессия генов может быть заблокирована путем вмешательства в нормальный путь передачи сигнала. Кроме того, мы также можем использовать различные методы для блокировки связывания рецепторов и лигандов и дальнейшего блокирования роли лигандов.
Терапия CAR-макрофагами (CAR-M)
В последние годы терапия химерными антигенными рецепторами (CAR) Т-клетками быстро развивается при лечении гематологических опухолей.В то же время макрофаги человека, которые модифицированы специфическим CAR для целевого антигена (например, CD19 и HER2) с целью распознавания опухолевых клеток и улучшения способности к фагоцитозу и презентации антигена, находятся в разработке в качестве иммунотерапии [191]. . О применении CAR-M-терапии при солидных опухолях впервые сообщили Klichinsky et al. в 2020 году [192]. Исследование показало, что CAR-M, нацеленный на HER2, значительно снижает опухолевую нагрузку, ингибирует метастазирование в легкие и продлевает время выживания в модели рака яичников на мышах [192].Более того, экспрессия структуры CAR может также способствовать поляризации ТАМ и даже обращать макрофаги М2 в макрофаги М1 [192, 193]. Кроме того, CAR-экспрессирующие макрофагальные клетки (CAR-iMac) на основе индуцированных плюрипотентных стволовых клеток (ИПСК) показали способность ингибировать рост опухолевых клеток как в гематологической, так и в модели рака яичников с подъемом экспрессии провоспалительных цитокинов М1 [193]. .
в 2020 году [192]. Исследование показало, что CAR-M, нацеленный на HER2, значительно снижает опухолевую нагрузку, ингибирует метастазирование в легкие и продлевает время выживания в модели рака яичников на мышах [192].Более того, экспрессия структуры CAR может также способствовать поляризации ТАМ и даже обращать макрофаги М2 в макрофаги М1 [192, 193]. Кроме того, CAR-экспрессирующие макрофагальные клетки (CAR-iMac) на основе индуцированных плюрипотентных стволовых клеток (ИПСК) показали способность ингибировать рост опухолевых клеток как в гематологической, так и в модели рака яичников с подъемом экспрессии провоспалительных цитокинов М1 [193]. .
Иммунные вакцины на основе ТАМ
В качестве новой стратегии противоопухолевые иммунные вакцины на основе макрофагов могут изменить форму ТМО и предотвратить рецидив опухоли после операции.Ван и др. разработали биологически слитую цитомембрану (FM), которая содержала как макрофаг M1, так и цитомембрану клеток опухоли молочной железы 4T1 [194]. С помощью вакцинных адъювантных цитозин-фосфат-гуанозиновых олигодезоксинуклеотидов (CpG ODN) вакцина успешно увеличивает количество провоспалительных макрофагов M1, способствует созреванию дендритных клеток и активирует Т-клетки памяти [194]. С другой стороны, комбинированная вакцина, состоящая из терапевтической вакцины и ингибитора рецептора ТАМ, обеспечивает новый способ лечения рака путем ингибирования активированных рецепторов ТАМ после лечения вакциной [191].
С помощью вакцинных адъювантных цитозин-фосфат-гуанозиновых олигодезоксинуклеотидов (CpG ODN) вакцина успешно увеличивает количество провоспалительных макрофагов M1, способствует созреванию дендритных клеток и активирует Т-клетки памяти [194]. С другой стороны, комбинированная вакцина, состоящая из терапевтической вакцины и ингибитора рецептора ТАМ, обеспечивает новый способ лечения рака путем ингибирования активированных рецепторов ТАМ после лечения вакциной [191].
ТАМ-нанобиотехнология
Нанобиотехнология, нацеленная на ТАМ, является еще одним многообещающим методом иммунотерапии. Наночастицы, нагруженные агонистами TLR, цитокинами, моноклональными антителами и оксидами металлов, достигли предпочтительных результатов в терапии рака [195, 196]. Доказана эффективность CAR-M в сочетании с наноносителем при солидных опухолях [197]. Канг и др. лечили моноциты или макрофаги с генами, кодирующими CAR и IFN-γ, через полимерные наноносители, что успешно приводило к фенотипическому переходу от М2 к М1 и усилению фагоцитоза макрофагов [197]. В модели рака молочной железы ТАМ были модифицированы наночастицами металлического органического каркаса на основе железа (Fe-MOF) с агентами, индуцирующими ферроптоз, что позволило ионам железа высвободиться в ТАМ и дополнительно усилить стресс, связанный с ферроптозом, в макрофагах, чтобы вызвать эффективную фенотипическую поляризацию M1. 195]. Наночастицы, покрытые ТАМ-мембраной, с конъюгированным фотосенсибилизатором (NPR@TAMM) и CSFR не только поглощают CSF1, полученный из опухоли, но также индуцируют трансформацию макрофагов M2 в M1 с помощью фотодинамической иммунотерапии [198].Кроме того, наночастицы, нагруженные агонистом TLR-7/TLR-8 резиквимодом (R848) и доксорубицином (DOX), вызывающим иммуногенную гибель клеток (ICD), или нагруженные куркумином (Cur) и байкалином (Bai), обладали удовлетворительной опухолевой цитотоксичностью при ТМЭ. значительно повысили эффективность иммунотерапии [199, 200].
В модели рака молочной железы ТАМ были модифицированы наночастицами металлического органического каркаса на основе железа (Fe-MOF) с агентами, индуцирующими ферроптоз, что позволило ионам железа высвободиться в ТАМ и дополнительно усилить стресс, связанный с ферроптозом, в макрофагах, чтобы вызвать эффективную фенотипическую поляризацию M1. 195]. Наночастицы, покрытые ТАМ-мембраной, с конъюгированным фотосенсибилизатором (NPR@TAMM) и CSFR не только поглощают CSF1, полученный из опухоли, но также индуцируют трансформацию макрофагов M2 в M1 с помощью фотодинамической иммунотерапии [198].Кроме того, наночастицы, нагруженные агонистом TLR-7/TLR-8 резиквимодом (R848) и доксорубицином (DOX), вызывающим иммуногенную гибель клеток (ICD), или нагруженные куркумином (Cur) и байкалином (Bai), обладали удовлетворительной опухолевой цитотоксичностью при ТМЭ. значительно повысили эффективность иммунотерапии [199, 200].
CDK12 способствует прогрессированию рака шейки матки за счет усиления инфильтрации макрофагов
Рак шейки матки (РШМ) является часто диагностируемым и основным фактором смерти онкологических больных при злокачественных новообразованиях женской репродуктивной системы. Циклинзависимая киназа 12 (CDK12), как CDK, связанная с транскрипцией, играет важную роль в поведении, способствующем развитию опухоли, тогда как основные механизмы CDK12 в прогрессировании РШМ до сих пор неясны. В этом отчете мы исследовали роль CDK12 в развитии рака шейки матки. Текущее исследование выявило CDK12 мРНК и экспрессию белка, значительно повышающую экспрессию у пациентов с РШМ. Повышенная экспрессия CDK12 была тесно связана с прогрессированием РШМ и неблагоприятным прогнозом. In vitro и функциональные эксперименты in vivo показали, что нокдаун CDK12 ингибирует пролиферацию раковых клеток и образование колоний и способствует апоптозу. Дальнейшие исследования показали, что CDK12 регулируют иммунную микросреду, способствуя прогрессированию клеток CC, способствуя инфильтрации макрофагов. Между тем, мы впервые продемонстрировали, что ядерный импорт CDK12 опосредуется TNPO1 и может быть новой терапевтической мишенью в онкологии.
Циклинзависимая киназа 12 (CDK12), как CDK, связанная с транскрипцией, играет важную роль в поведении, способствующем развитию опухоли, тогда как основные механизмы CDK12 в прогрессировании РШМ до сих пор неясны. В этом отчете мы исследовали роль CDK12 в развитии рака шейки матки. Текущее исследование выявило CDK12 мРНК и экспрессию белка, значительно повышающую экспрессию у пациентов с РШМ. Повышенная экспрессия CDK12 была тесно связана с прогрессированием РШМ и неблагоприятным прогнозом. In vitro и функциональные эксперименты in vivo показали, что нокдаун CDK12 ингибирует пролиферацию раковых клеток и образование колоний и способствует апоптозу. Дальнейшие исследования показали, что CDK12 регулируют иммунную микросреду, способствуя прогрессированию клеток CC, способствуя инфильтрации макрофагов. Между тем, мы впервые продемонстрировали, что ядерный импорт CDK12 опосредуется TNPO1 и может быть новой терапевтической мишенью в онкологии. В совокупности это исследование указало на потенциал CDK12 в качестве новой терапевтической мишени для ограничения пролиферации CC и процесса клеточного цикла за счет стимулирования инфильтрации макрофагов.
В совокупности это исследование указало на потенциал CDK12 в качестве новой терапевтической мишени для ограничения пролиферации CC и процесса клеточного цикла за счет стимулирования инфильтрации макрофагов.
1. Введение
Заболеваемость раком шейки матки (РШМ) становится одним из наиболее распространенных злокачественных новообразований женской репродуктивной системы, особенно в странах с более низким ИЧР (индексом человеческого развития), несмотря на прогресс в профилактических вакцинах [1]. Между тем, ранний рак шейки матки можно лечить с помощью хирургического вмешательства или лучевой терапии, но на поздних стадиях РШМ его эффективность остается низкой [2].Следовательно, более глубокое понимание молекулярного механизма агрессии СС будет иметь решающее значение для разработки эффективных стратегий лечения СС.
Циклинзависимые киназы (ЦДК) являются важными регуляторами биологического процесса клеточного цикла [3]. CDK обычно делятся на два подсемейства: такие как CDK2 и CDK4, которые непосредственно регулируют ход клеточного цикла. Другие CDK связаны с транскрипцией генов, такие как CDK12 и CDK13, которые в основном участвуют в посттранскрипционном процессинге мРНК [4].Масса исследований свидетельствует о подавлении или повышении экспрессии CDK при различных видах рака [5]. В этом контексте нарушение работы нескольких связанных с транскрипцией CDK было связано с онкогенезом и прогрессированием, подтверждая правдоподобные корреляции посттранскрипционного процессинга мРНК с этиопатогенезом рака [6]. Таким образом, CDK широко тестировались в качестве потенциальных мишеней для терапии рака.
Другие CDK связаны с транскрипцией генов, такие как CDK12 и CDK13, которые в основном участвуют в посттранскрипционном процессинге мРНК [4].Масса исследований свидетельствует о подавлении или повышении экспрессии CDK при различных видах рака [5]. В этом контексте нарушение работы нескольких связанных с транскрипцией CDK было связано с онкогенезом и прогрессированием, подтверждая правдоподобные корреляции посттранскрипционного процессинга мРНК с этиопатогенезом рака [6]. Таким образом, CDK широко тестировались в качестве потенциальных мишеней для терапии рака.
Циклинзависимая киназа 12 (CDK12), как CDK, связанная с транскрипцией, образует комплексы с циклином K, чтобы модулировать удлинение транскрипции гена посредством фосфорилирования C-концевого домена (CTD) РНК-полимеразы II (RNA Pol II) в Ser2 [7 ]. In vitro , CDK12 также потенциально модулирует инициацию транскрипции путем фосфорилирования РНК Pol II по Ser5 и принимает участие в ряде клеточных биологических процессов, включая ответ на повреждение ДНК (DDR), пролиферацию клетки, сплайсинг мРНК-предшественника и процесс претранскрипционной мРНК [8, 9]. Более того, CDK12 также выполняет важные функции в регуляции процессов клеточного цикла. Предыдущие исследования показали, что при некоторых злокачественных новообразованиях, включая рак яичников, молочной железы и предстательной железы, наблюдались высокочастотные мутации и амплификация экспрессии CDK12 [10, 11].В соответствии с его функцией в поддержании стабильности генома и регуляции клеточного цикла потеря или сверхэкспрессия CDK12 связана с прогрессированием и метастазированием рака [12].
Более того, CDK12 также выполняет важные функции в регуляции процессов клеточного цикла. Предыдущие исследования показали, что при некоторых злокачественных новообразованиях, включая рак яичников, молочной железы и предстательной железы, наблюдались высокочастотные мутации и амплификация экспрессии CDK12 [10, 11].В соответствии с его функцией в поддержании стабильности генома и регуляции клеточного цикла потеря или сверхэкспрессия CDK12 связана с прогрессированием и метастазированием рака [12].
Микроокружение опухоли (ММО) как сложная экосистема содержит мезенхимальные, опухолевые ткани и воспалительные клетки и способствует росту, инвазии и метастазированию раковых клеток [13]. Воспалительные клетки как основной компонент ОМО, среди которых опухолеассоциированные макрофаги (ТАМ), играют важную роль в онкогенезе и прогрессировании [14].ТАМ способствуют выживанию и пролиферации опухолевых клеток путем высвобождения тромбоцитарного провоспалительного фактора роста (PDGF), стимулируют ангиогенез опухоли путем высвобождения фактора роста эндотелия сосудов (VEGF) и способствуют метастазированию опухоли, индуцируя ядерный фактор каппа B (NF- κ В) путь [15].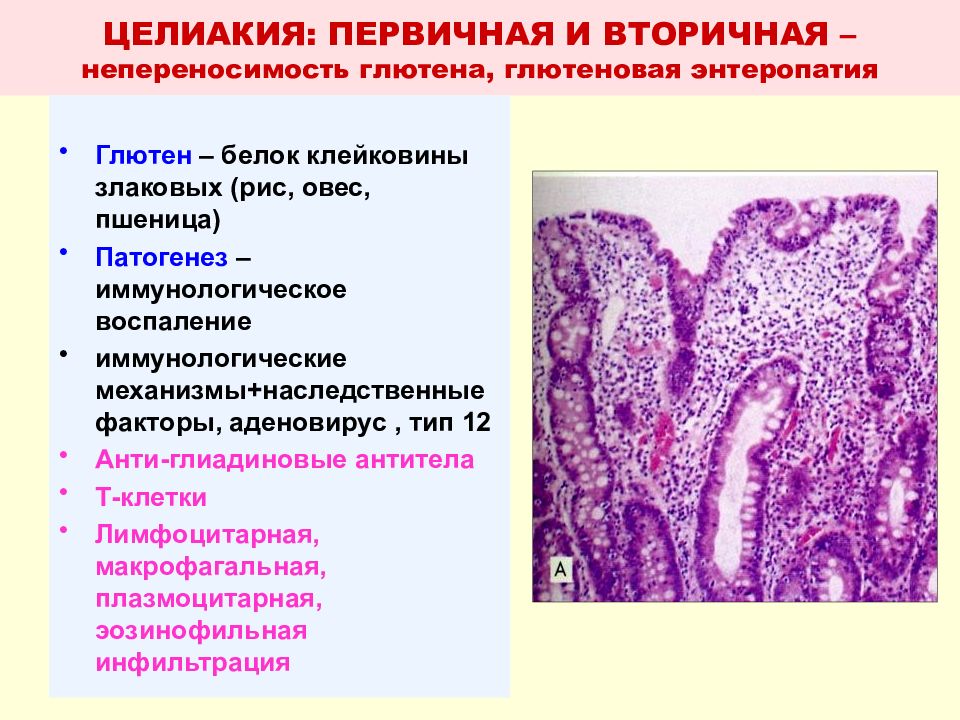 Таким образом, нацеливание на ТАМ имеет огромную возможность стать многообещающей и эффективной терапевтической стратегией в сочетании с традиционной терапией и привлекло всестороннее внимание к нацеливанию на ТАМ для иммунотерапии рака, которая направлена на иммунокомпетентность организма [16].
Таким образом, нацеливание на ТАМ имеет огромную возможность стать многообещающей и эффективной терапевтической стратегией в сочетании с традиционной терапией и привлекло всестороннее внимание к нацеливанию на ТАМ для иммунотерапии рака, которая направлена на иммунокомпетентность организма [16].
В настоящем исследовании экспрессия CDK12 была значительно повышена при раке шейки матки по сравнению с нормальной шейкой матки и имела тенденцию к неблагоприятному прогнозу. Нокдаун CDK12 ингибировал пролиферацию CC-клеток, индуцировал остановку клеточного цикла на G2/M и более высокий коэффициент апоптоза CC-клеток. Дальнейшие исследования показали, что CDK12 способствует инфильтрации макрофагов и регулирует иммунное микроокружение в клетках рака шейки матки. Более того, наши результаты выявили новый молекулярный механизм, при котором импорт CDK12 в ядро опосредуется TNPO1 и может быть новой терапевтической мишенью в онкологии.
2. Метод
2.1.
 Образцы тканей CC человека
Образцы тканей CC человекаВ нашем исследовании из Шестой больницы Шанхая были взяты свежезамороженные ткани, из которых было получено 20 образцов тканей CC и 20 образцов эпителиальной ткани шейки матки. У всех пациентов диагноз РХ был подтвержден патологоанатомом. Никому из больных не проводилась химиолучевая терапия до оперативного лечения. Это исследование было санкционировано муниципальным правительством Шанхая, и перед этим исследованием всем пациентам было предоставлено информированное согласие.
2.2. Культура клеток
Рак шейки матки человека Клетки HeLa и Siha были сохранены в Шанхайском институте рака, больнице Жэнь Цзи, Медицинской школе Шанхайского университета Цзяо Тонг. Клетки HeLa и Siha поддерживали в среде DMEM (Gibco) и добавляли 10% фетальной телячьей сыворотки и (об/об) пен/стрептококковые антибиотики. Все они инкубировались при 37°C и атмосфере, содержащей 5% CO 2 .
2.3. Иммуногистохимическое окрашивание
Образцы ткани СС, нормальной эпителиальной ткани шейки матки и ксенотрансплантата мышей помещали в парафин для иммуногистохимии. Окрашивание ИГХ было показано в предыдущем исследовании [12]. Используемым первичным антителом было анти-CDK12 (разведение 1 : 1000, 11973 с, CST). На последнем этапе все образцы были исследованы и сфотографированы под микроскопом. Интенсивность окрашивания оценивали следующим образом: заметное окрашивание оценивали в 0 баллов, слабое коричневое окрашивание — в 1 балл, коричневое окрашивание — в 2 балла, темно-коричневое окрашивание — в 3 балла, а степень окрашивания оценивали как 0 (<10%), 1 (10–40 баллов). %), 2 (41–70%) или 3 (>70%). Образцы с итоговой оценкой не более 3 были определены как «низкая экспрессия».Эти баллы были оценены двумя патологоанатомами независимо друг от друга слепым методом.
Окрашивание ИГХ было показано в предыдущем исследовании [12]. Используемым первичным антителом было анти-CDK12 (разведение 1 : 1000, 11973 с, CST). На последнем этапе все образцы были исследованы и сфотографированы под микроскопом. Интенсивность окрашивания оценивали следующим образом: заметное окрашивание оценивали в 0 баллов, слабое коричневое окрашивание — в 1 балл, коричневое окрашивание — в 2 балла, темно-коричневое окрашивание — в 3 балла, а степень окрашивания оценивали как 0 (<10%), 1 (10–40 баллов). %), 2 (41–70%) или 3 (>70%). Образцы с итоговой оценкой не более 3 были определены как «низкая экспрессия».Эти баллы были оценены двумя патологоанатомами независимо друг от друга слепым методом.
2.4. Количественная ПЦР в реальном времени
Суммарную мРНК выделяли из клеток CC с использованием реагента TRIzol (TaKaRa) в соответствии с рабочими спецификациями. Количественную ПЦР в реальном времени проводили с использованием SYBR Premix Ex Taq (TaKaRa) на системе ПЦР в реальном времени 7500 (Applied Biosystems, Inc. , США). Относительные уровни мРНК каждого гена нормализовали к экспрессии эталонного гена 18S. Последовательности праймеров, использованные для обнаружения CDK12 и 18s РНК, были следующими:
, США). Относительные уровни мРНК каждого гена нормализовали к экспрессии эталонного гена 18S. Последовательности праймеров, использованные для обнаружения CDK12 и 18s РНК, были следующими:
CDK12 прямой 5-CTAACAGCAGAGAGCGTCACC-3,
CDK12 обратный 5-AAAGGTTTGATAACTGTGCCCA-3;
18 с вперед 5-TGCGAGTACTCAACACCAACA-3,
18 с назад 5-GCATATCTTCGGCCCACA-3;
Формула использовалась для количественного определения относительных уровней экспрессии целевого гена для статистического анализа.
2.5. Нокдаун генов, опосредованный короткой шпилькой РНК
кшРНК, нацеленных на разные гены, были приобретены у Gene Pharma (Шанхай, Китай). Раковые клетки трансдуцировали рекомбинантными единицами, трансдуцирующими лентивирус, в присутствии 5 мк мкг/мл полибрена (H9268, Sigma). Для работы, связанной с лентивирусным вектором, клетки (через 48 часов после трансдукции) отбирали и поддерживали в 2 мк мкг/мл пуромицина. Эффективность нокдауна проверяли с помощью количественной ПЦР в реальном времени и вестерн-блоттинга.Идентификаторы клонов для кшРНК CDK12 были следующими:
Эффективность нокдауна проверяли с помощью количественной ПЦР в реальном времени и вестерн-блоттинга.Идентификаторы клонов для кшРНК CDK12 были следующими:
sh2,5-GATCCGCCTTCAAACTAGACCGAAGGTTCAAGAGACCTTCGGTCTAGTTTTGAAGGCTTTTTTG-3,
sh3,5GATCCGGAGAAGACCAGGAAAGAACGTTCAAGAGACGTTCTTTCCTGGTCTTCTCCTTTTTTG-3.
2.6. Вестерн-блоттинг
Лизаты белков цельных клеток готовили в соответствии с обычными протоколами, а вестерн-блоттинг проводили, как описано ранее [13]. Используемые антитела представляли собой анти-CDK12 (11973s, CST), анти-GAPDH (60004-1-Ig, Proteintech) и анти-TNPO1 (ab10303, Abcam).Для иммунопреципитации экстракты инкубировали с белком A/G Dynabeads (Bimake). После этого шарики трижды промывали фосфатно-солевым буфером, ресуспендировали в однократной загрузке SDS белком и кипятили в течение 15 минут. Фракционирование белков субклеточной ядерной цитоплазмы проводили с использованием реагентов для выделения ядер и цитоплазмы NE-PER (Thermo Fisher Scientific) в соответствии с протоколом, предоставленным производителем. Вторичные антитела использовали при 1 : 5000, включая антимышиные (G-21040, Invitrogen) и антикроличьи (G-21234, Invitrogen) соответственно.
Вторичные антитела использовали при 1 : 5000, включая антимышиные (G-21040, Invitrogen) и антикроличьи (G-21234, Invitrogen) соответственно.
2.7. Анализ жизнеспособности клеток
Клетки HeLa и Siha высевали в 96-луночные планшеты со средой 200 мкл л (2000 клеток/лунку) и культивировали при 37°C. После добавления 10% реагента Cell Counting Kit-8 (CCK-8, Dojindo), соответственно, клетки культивировали при 37°C в течение 1 часа. Жизнеспособность клеток измеряли при 450 нм с помощью устройства для считывания микропланшетов Power Wave XS (BioTek, Winooski).
2.8. Анализ образования колоний
После трансфекции клеток sh- CDK12 -1, sh- CDK12 -2 или shNC HeLa и Siha и их контрольные клетки помещали в 6-луночные планшеты ().Через 10-14 дней колонии отмывали PBS и окрашивали 0,05% кристаллическим фиолетовым в течение 15 мин. Количество видимых колоний подсчитывали под микроскопом.
2.9. Анализ проточной цитометрии клеточного апоптоза Клетки
HeLa и Siha, трансфицированные sh- CDK12 -1, sh- CDK12 -2 или shNC, культивировали в условиях депривации сыворотки в течение 24 часов для оценки апоптоза. Используя 0,25% трипсин, клетки ресуспендировали (100 мкл л) для окрашивания аннексином V йодидом пропидия (PI) и аннексином V-FITC (BD Pharmingen, США).Процент клеток с помощью анализатора клеток LSRFortessa (BD Biosciences) и результаты рассчитывали с помощью программного обеспечения FlowJo 10.0.
2.10. Анализ клеточного цикла Клетки
HeLa и Siha, трансфицированные sh- CDK12 -1, sh- CDK12 -2 или shNC, культивировали в условиях депривации сыворотки в течение 48 часов для оценки клеточного цикла. Клетки фиксировали в 75% ледяном этиловом спирте при 4°С в течение 12 часов. После этого клетки суспендировали в PBS, содержащем 10 мг/л РНКазы А и 50 мкг/мл мкг/мл PI, и инкубировали в темноте при комнатной температуре в течение 30 мин.Содержание ДНК исследовали с помощью анализатора клеток LSRFortessa (BD Biosciences), а результаты рассчитывали с помощью программного обеспечения Modfit LT 5.1.
2.11. Модель ксенотрансплантата мышей
Для создания модели ксенотрансплантата клетки HeLa и Siha, трансфицированные sh- CDK12 -1 или shNC в 150 мкл мкл PBS, вводили подкожно в паховую область каждому самцу мышей BALB/c ( в возрасте 5-6 недель). Объем опухоли измеряли штангенциркулем каждые 6 дней. Мышей умерщвляли через 28 дней после инъекции, а опухоли ксенотрансплантата вырезали для измерения объема и веса.Все животные получали гуманный уход в соответствии с местными или национальными требованиями по уходу и использованию лабораторных животных.
2.12. Окрашивание Edu Иммунофлуоресценция и конфокальная микроскопия
Клетки HeLa и Siha, трансфицированные sh- CDK12 -1, sh- CDK12 -2 или shNC, высевали на 8-луночные предметные стекла, затем инкубировали с сывороткой. — свободная среда, содержащая указанные дозы гераниина в течение 24 часов и совместно обработанная 100 мкг л EdU (50 мкг моль/л) в течение 2 часов.Клетки фиксировали 4% параформальдегидом при 37°C в течение 15 мин и промывали PBS, затем пермеабилизировали 0,5% раствором Triton X-100. Флуоресцентные изображения получали с помощью конфокального микроскопа (Carl Zeiss, Германия) в соответствии с рекомендациями производителя.
2.13. Статистический анализ
Данные представлены в формате . Программное обеспечение SPSS (версия 19.0; IBM Corporation) и программное обеспечение GraphPad Prism 7 (La Jolla, CA) использовали для статистического анализа. Критерий Стьюдента применялся для анализа двух групп данных.Однофакторный дисперсионный анализ использовался для включения нескольких групп. Значения считались статистически значимыми.
3. Результаты
3.1. CDK12 способствует злокачественной трансформации и плохому прогнозу при раке шейки матки
Генная мутация и амплификация CDK12 были обнаружены при различных типах злокачественных новообразований путем анализа баз данных Атласа генома рака (TCGA), таких как амплификация CDK12 в шейке матки. рак (СС), что позволило предположить, что CDK12 обладает некоторыми свойствами онкогена (рис. 1(а)).Следует отметить, что амплификация экспрессии CDK12 показала плохой прогноз у пациентов с СС (рис. 1(b)). Чтобы дополнительно раскрыть дисрегуляцию CDK12 , связанную с прогрессированием РШМ, мы первоначально опросили профили экспрессии генов CDK12 в различных наборах данных Gene Expression Omnibus (GEO) (GSE6791 и GSE7803), которые все содержат нормальную шейку матки и ткани РХ. Результаты анализа показали, что экспрессия мРНК CDK12 была заметно увеличена при РШМ (рис. 1(c) и 1(d)).Кроме того, CIN (цервикальная интраэпителиальная неоплазия) является заметным предраковым поражением CC, особенно HSIL (плоскоклеточное интраэпителиальное поражение высокой степени). Интересно, что мы указали, что экспрессия CDK12 была заметно увеличена в CC по сравнению с HSIL в GSE7803 (рис. 1 (d)). Чтобы оценить уровень белка CDK12 в образцах ткани РШМ, мы провели иммуногистохимическое окрашивание серии образцов пациентов, в результате чего было получено 20 образцов ткани РШМ и 20 образцов нормальной ткани шейки матки.Результаты анализа показали, что уровень белка CDK12 был заметно повышен в СС по сравнению с нормальными образцами ткани шейки матки (рис. 1(e)). В совокупности мы предположили, что CDK12 являются специфическим онкогеном, избирательно способствующим прогрессированию РШМ.
3.2. CDK12 регулирует опухолевой генез и пролиферацию In Vitro
Для выяснения функций CDK12 при прогрессировании РШМ мы сначала отобрали клеточные линии HeLa и Siha CC, которые с относительно более высокими уровнями экспрессии CDK12 (дополнение к рис. 1(а) ).Затем мы установили нокдаун клеток CDK12 с помощью РНК с короткой шпилькой (shRNA), нацеленной на CDK12 . Между тем, мы также установили сверхэкспрессию клеток CDK12 с помощью трансфекции, опосредованной лентивирусом. Количественная ПЦР в реальном времени и вестерн-блоттинг подтвердили эффективность нокдауна и сверхэкспрессии CDK12 в клетках Hela и Siha (дополнение к рисунку 1(b, c)). В присутствии 10% FBS анализ показал, что нокдаун CDK12 заметно снижает жизнеспособность клеток HeLa и Siha по сравнению с shNC (рис. 2(а)).Следуя этим выводам, сверхэкспрессия CDK12 заметно повышала жизнеспособность клеток HeLa и Siha (рис. 2(b)). Чтобы дополнительно проверить роль CDK12 в пролиферации клеток CC, мы провели анализ образования колоний. В соответствии с этими выводами образование колоний подавлялось нокдауном CDK12 и стимулировалось сверхэкспрессией CDK12 (рис. 2(c) и 2(d)). Кроме того, мы провели анализ окрашивания EdU для обнаружения репликации ДНК.Наши результаты также показали, что репликация ДНК была подавлена нокдауном CDK12 (рис. 2(e)). В совокупности эти результаты показали, что CDK12 глубоко вовлечен в стимулирование пролиферации CC.
3.3. Нокдаун CDK12 регулирует клеточный цикл и индуцирует апоптоз In Vitro
CDK12 как важный фактор, регулирующий процесс клеточного цикла, обычно участвует в росте раковых клеток. Поэтому мы использовали проточную цитометрию для изучения влияния CDK12 на прогрессирование клеточного цикла рака шейки матки.Результаты анализа клеточного цикла FACS показали, что нокдаун CDK12 привел к увеличению доли клеток HeLa и Siha в фазе G2/M и значительно увеличился по сравнению с группой shNC (рис. 3(а)). Кроме того, мы также провели окрашивание аннексином V (V) и йодидом пропидия (PI) и анализ проточной цитометрии для определения соотношения апоптотических и мертвых клеток соответственно. Эти результаты анализа показали, что коэффициент апоптоза sh CDK12 -1 и sh CDK12 -2 в клеточной линии HeLa и Siha был заметно выше, чем в группе shNC (рис. 3(b)).Предыдущее открытие ковалентного ингибитора CDK12 THZ531 выявило последовательное ингибирование активности киназы CDK12 и подавление пролиферации раковых клеток [17]. Чтобы оценить, обладает ли THZ531 аналогичной противораковой активностью, мы обработали клетки HeLa и Siha 200 нМ THZ531 в течение 48 часов. Анализ клеточного цикла FACS после обработки THZ531 показал резкое увеличение доли клеток, демонстрирующих содержание G2/M (рис. 3(c)). Для дальнейшего исследования мы провели окрашивание аннексином V/PI, чтобы определить влияние THZ531 на клетки рака шейки матки.При 200 нМ THZ531 наблюдалось резкое увеличение доли апоптотических клеток при обработке высокими дозами THZ531 по сравнению с контролем ДМСО в эксперименте (рис. 3(d)). Эти результаты показали, что нокдаун CDK12 значительно ингибирует клеточный цикл и индуцирует апоптоз клеток рака шейки матки.
3.4. Нокдаун экспрессии CDK12 ингибирует опухолевой генез In Vivo
Для дальнейшего анализа влияния нокдауна CDK12 на пролиферацию клеток CC были проведены исследования in vivo с помощью подкожного введения ксенотрансплантата мышам с sh CDK12 в клетках HeLa и Siha.Результаты показали, что в группе sh CDK12 значительно уменьшилась опухолевая масса, что оценивалось по измерениям веса и объема опухоли в клетках HeLa и Siha (рис. 4(a) и 4(b)). Кроме того, результаты иммуногистохимического окрашивания показали, что интенсивность иммунного окрашивания PCNA и Ki-67, маркеров клеточной пролиферации, была значительно снижена в группе sh CDK12 по сравнению с соответствующей группой shNC в клетках HeLa и Siha (рис. 4(c)) . В совокупности эти результаты показали, что нокдаун CDK12 ингибирует онкогенез и рост при РШМ.
3.5. CDK12 усиливает инфильтрацию макрофагов для стимулирования пролиферации CC
Показав, что CDK12 выполняет важные функции в стимулировании свойств пролиферации CC, мы намеревались дополнительно изучить взаимосвязь молекулярного механизма с функцией CDK12 . Чтобы исследовать потенциальный механизм действия CDK12 при раке шейки матки, мы провели высокопроизводительный анализ секвенирования транскриптома (RNA-seq) для сравнения транскриптома двух групп: групп shNC и sh CDK12 соответственно (рис. 5(a) и 5). (б)).Мы выполнили GSEA (анализ обогащения набора генов) для анализа дифференцированных генов в группах с высокой и низкой экспрессией CDK12 в базе данных RNA-seq. Результаты анализа показали, что высокая экспрессия CDK12 была обогащена некоторыми путями иммунно-воспалительного ответа, такими как TNFA_SIGNALING_VIA_NFKB и HALLMARK_INFLAMMATORY_RESPONSE (рис. 5(c)). CDK12 может принимать участие в регуляции иммунного микроокружения клеток РШМ. Поэтому мы исследовали корреляцию экспрессии CDK12 и дифференциальной распространенности инфильтрации иммунных клеток с использованием баз данных TCGA.Базу данных Timer использовали для расчета корреляции экспрессии CDK12 с восемью типами инфильтрации иммунных клеток в тканях пациентов с CC. Результаты анализа показали, что экспрессия CDK12 значимо положительно коррелирует с инфильтрацией макрофагов M0 (Фигура 5(d)). Кроме того, результаты иммуногистохимического окрашивания показали, что интенсивность иммуноокрашивания макрофагов была значительно снижена в группе sh CDK12 по сравнению с группой shNC в подкожном ксенотрансплантате клеток HeLa и Siha (рис. 5 (e)).В совокупности эти результаты показали, что CDK12 способствует пролиферации СС, регулируя иммунное микроокружение, которое зависит от усиления инфильтрации макрофагов.
3.6. Кариоферин TNPO1 модулирует ядерный импорт CDK12 в CC-клетках
CDK12, как серин/треонинкиназа, коррелирующая с транскрипцией, играет важную роль в регуляции экспрессии генов посредством стимулирования фосфорилирования РНК POL II и индукции удлинения транскрипции.Для обеспечения точной функции CDK12, связанной с транскрипцией генов, пространственное распределение белка CDK12 требует опосредования белка кариоферина β (Kap β s). Поэтому мы дополнительно исследовали потенциальный белок кариоферин β (Kap β s), участвующий в ядерно-цитоплазматическом транспорте CDK12. Мы обнаружили, что CDK12, по крайней мере, содержит консенсусную последовательность мотива PY-NLS (пролин-тирозиновая аминокислота), которая была распознана транспортином 1 ( TNPO1 ), путем анализа канонической последовательности CDK12 на основе базы данных UniProt.Затем мы отметили наличие строго консервативного аргинина и нижестоящего мотива PY-NLS в аминокислоте CDK12 295-315, и этот мотив высоко консервативен в различных сплайсосомах Homo sapiens и различных видов млекопитающих (рис. 6(а)). Поэтому мы провели коиммунопреципитацию против CDK12, связанного с TNPO1, в клетках HeLa и Siha (рис. 6(b)). Между тем, мы провели вестерн-блоттинг на отдельных ядерных и цитоплазматических фракциях клеток HeLa и Siha и дополнительно представили, что ядерный импорт CDK12 уменьшался, когда TNPO1 был нокдаун (рис. 6 (c)).Более того, учитывая, что импорт CDK12 в ядро регулируется TNPO1, мы использовали пептид-ингибитор, специфичный для TNPO1 (M9M). Вестерн-блоттинг на отдельных ядерных и цитоплазматических фракциях клеток HeLa и Siha, а также показал, что ядерный импорт CDK12 уменьшался при экспрессии конструкции M9M (рис. 6(c)). Чтобы дополнительно подтвердить, что импорт CDK12 в ядро опосредован TNPO1, мы провели иммунофлуоресцентный анализ этих клеток, чтобы дополнительно показать, что CDK12 разбросала локализацию в ядре и цитоплазме, а ядерная локализация эндогенного CDK12 была заметно нарушена, когда TNPO1 был нокдаун ( Рисунок 6(г)).
4. Обсуждение
Циклинзависимые киназы (CDK) являются важными регуляторами процесса клеточного цикла и транскрипции генов. Поскольку нарушение регуляции CDK часто происходит во время онкогенеза, CDK превратились в важные белки-мишени для лечения рака. В качестве CDK, связанных с транскрипцией, недавно сообщалось о нарушении регуляции гена CDK12 при различных типах злокачественных новообразований, что способствует прогрессированию и агрессивности рака.Целью этого исследования является изучение точных функций и основных механизмов CDK12 для онкогенеза и пролиферации при раке шейки матки.
Мутации, амплификации, делеции или слияние гена CDK12 недавно были зарегистрированы при различных типах рака, например, мутации с потерей функции геномных коррелируют с тандемными дупликациями (TD) и гомологичной рекомбинацией (HR) при высокозлокачественной серозной карциноме яичников и раке предстательной железы, что позволяет предположить, что CDK12 является супрессором опухоли.Напротив, наиболее распространенной геномной модификацией CDK12 являются амплификации. Сверхэкспрессия CDK12 обладает некоторыми свойствами онкогена в других опухолях и способствует пролиферации HER2 -положительного рака молочной железы, что приводит к метастазированию и плохому прогнозу. В этом исследовании широко распространенный компьютерный биоинформационный анализ из некоторых независимых баз данных и клинических образцов тканей пациентов показывает, что уровень гена и белка CDK12 значительно увеличивается при раке шейки матки, а сверхэкспрессия CDK12 тесно коррелирует с неблагоприятным прогнозом для пациентов, страдающих от рак шейки матки.Поэтому мы считаем, что CDK12 может быть плохим прогностическим маркером рака шейки матки.
Известно, что нокдаун CDK12 может ингибировать прогрессирование клеточного цикла рака пищевода и легкого [18, 19]. Предыдущие исследования обнаружили пик накопления CDK12 в ранней фазе G1, и он сыграл решающую роль в переходе G1 в фазу S [20]. Анализ клеточного цикла показал, что нокдаун CDK12 снижает прогрессирование фазы G1/S, что является важным процессом для репликации ДНК в клеточном цикле [21].Точно так же в настоящем исследовании мы обнаружили, что нокдаун CDK12 ингибирует переход от G1 к S-фазе и репликацию ДНК в клетках рака шейки матки. Было показано, что подавление CDK12 или THZ531, ингибитора CDK12, ингибирует пролиферацию клеток и вызывает апоптоз в раковых клетках [22]. В настоящем исследовании мы также обнаружили, что нокдаун CDK12 с помощью обработки shRNA или THZ531 ингибирует пролиферацию клеток, подавляет образование колоний и способствует их апоптозу в клетках рака шейки матки.Сообщалось, что
ТАМ, как разновидность миелоидных иммунных клеток, имеют важное прогностическое значение при злокачественных опухолях человека [16]. Текущие исследования показали, что повышенная инфильтрация в TME связана с худшим прогнозом для пациентов с раком легких и молочной железы [23, 24]. Помимо рака легкого и молочной железы, повышенное накопление ТАМ при раке желудка и множественной миеломе также связано с плохим прогнозом [25]. CDK12 способствует регуляции онкогенов и биологических путей, таких как гены, связанные с суперэнхансерами (SE-), и путь NF- κ B, который участвует в гомеостатическом контроле иммунной системы [26].Между тем, потеря функции CDK12 является общей корреляцией с фенотипом TD и увеличивает инфильтрацию Т-лимфоцитов, что приводит к сенсибилизации раковых клеток к некоторым ингибиторам иммунных контрольных точек, таким как антипрограммированная гибель клеток-1 (PD-1) в простате. и рак яичников [27, 28]. В настоящем исследовании анализ обогащения набора генов в клетках рака шейки матки показал, что измененная экспрессия генов была обогащена в пути NF- κ B и путях иммунно-воспалительного ответа.Более того, сверхэкспрессия CDK12 усиливает инфильтрацию ТАМ методами иммуногистохимии и профилирования экспрессии генов. Эти результаты показали, что CDK12 может участвовать в регуляции TME в клетках CC.
CDK12 , который представляет собой CDK, связанный с транскрипцией, отвечает за инициацию транскрипции и удлинение гена посредством фосфорилирования CTD РНКП II в Ser2 и Ser5. CDK12 также регулирует экспрессию генов DDR и репликации ДНК, которые участвуют в клеточном цикле и стабильности генома, участвуя в сплайсинге и трансляции РНК [29].CDK12 представляет собой высокомолекулярную субстанцию, состоящую из 1490 аминокислот с массой 164 кДа [30]. Для обеспечения точной функции CDK12, связанной с транскрипцией генов, для ядерно-цитоплазматического транспорта белка CDK12 требуется белок кариоферин β (Kap β s). В настоящем исследовании наши результаты вестерн-блоттинга и иммунофлуоресценции впервые выявили новый молекулярный механизм ядерно-цитоплазматического транспорта CDK12, который модулируется TNPO1. TNPO1 опосредует ядерный импорт CDK12, а затем обеспечивает CDK12 выполнение своей функции регуляции транскрипции генов.
5. Заключение
В настоящем исследовании наши результаты показали, что избыточная экспрессия CDK12 связана с прогрессированием РШМ и неблагоприятным прогнозом. Нокдаун CDK12 ингибирует процесс клеточного цикла и пролиферацию клеток рака шейки матки. Результаты анализа GSEA дают представление о роли CDK12 в модуляции иммунной микросреды. Мы также обнаружили, что ядерный импорт CDK12 опосредован TNPO1. Эти результаты убедительно свидетельствуют о том, что CDK12 является потенциальной мишенью иммунотерапии при раке шейки матки.
Доступность данных
Данные, использованные для подтверждения результатов этого исследования, были предоставлены Yincheng Teng по лицензии и поэтому не могут быть размещены в свободном доступе. Запросы на доступ к этим данным должны рассматриваться соответствующим автором.
Конфликт интересов
Авторы заявляют, что у них нет конкурирующих интересов с содержанием этой статьи.
Вклад авторов
Yincheng Teng, Bikang Yang и Jing Chen разработали исследование.Цзин Чен и Бикан Ян провели эксперименты. Наконец, Бикан Ян написал рукопись. Все авторы прочитали и одобрили окончательный вариант рукописи. Бикан Ян и Цзин Чен внесли равный вклад в эту работу.
Благодарности
Это исследование было поддержано грантами Национального фонда естественных наук Китая (№: 81974406).
Дополнительные материалы
Дополнительные материалы На рисунке 1 показан уровень экспрессии мРНК CDK12 в клеточных линиях CC и эффективность интерференции или избыточной экспрессии CDK12. (дополнительные материалы). , R Schwarcz
10 июля 1989 г. · Исследование мозга · M Sawada T Marunouchi
1 августа 1986 г. · Журнал экспериментальной медицины · D Giulian L B Lachman
1 октября 1983 г. · Journal of Clinical Pathology · RI Harris J Stuart
5
5
5 28 апреля 1995 г. · Neuroscience Letters · T YoshidaS Hirai
1 июля 1994 г. · Journal of Comparative Pathology · M Hewicker-Trautwein, G Schultheis
1 декабря 1996 г. · The Biochemical Journal · MP Heyes S P Markey
4 сентября, 1998 · Исследования в области педиатрии · H KazdaD Walker
20 октября 1998 г. · Семинары по детской неврологии · HC Kinney, SA Back
22 декабря 1999 г. · Американский журнал акушерства и гинекологии · T NichollsD W Walker
12 апреля 2000 г. · Европейский журнал медицинской химии · TW Stone
29 января 2002 г. · Аналитическая биохимия · GA SmytheD W Walker
29 марта 2002 г. · Обзоры исследований умственной отсталости и нарушений развития · Олалан Лейфманн
19 июля 2002 г. · Журнал нейрохимии · Томоюки Кита С. П. Маркей
6 ноября 2002 г. · Нейропатология: Официальный журнал Японского общества невропатологии · Паям Резайе, Эндрю Дин
3 октября 2003 г. · Исследования сердечно-сосудистой системы · Самули Рунио Hallman
20 февраля 2004 г. · Pediatric Research · Edwin YanDavid Walker
26 марта 2004 г. · Glia · Mark A Petersen, Michael E Dailey
27 января 2005 г. · American Journal of Obstetrics and Gynecology · Ursula ManuelpillaiDavid0 W Walker 8 февраля 2005 г. · Семинары по перинатологии · Стивен А. Бэк, Скотт А. Ривкис
9 апреля 2005 г. · Журнал эндокринологии · L McClureD J Phillips
28 мая 2005 г. · The American Journal of Pathology · Samuli RouniojaMikko Hallman
29 июля 2005 г. · The Journal of Neuroscience: официальный журнал Общества нейробиологов · W Shawn CarbonellJames W Mandell
6 декабря 2005 г. · The European Journal of Neuroscience · HB StolpN R Saunders
февраля 24, 2006 · Детские исследования · дедиатрические исследования · Кэтрин I Roussetelie Saliba
16 мая 2006 г. · Журнал общества для гинекологического исследования · ILIAS Nitsostimothy JM MOSS
Цитаты
13 августа 2011 г. · Журнал нервной передачи · Эрин К. Стаховски, Роберт Шварц
24 июля 2013 г. · Исследования в области педиатрии · Lorelei PrattG Miller Jonakait
30 октября 2009 г. · Детская патология и патология развития: официальный журнал Общества детской патологии и Общества детской патологии · Линда М. ErnstMichal Elovitz
18 декабря 2013 г. · Педиатрические исследования · Карина Маллард Алистер Ян Ганн
31 июля 2013 г. · Журнал педиатрии · Джоанна C HartemanLinda S de Vries
17 сентября 2015 г. · Исследования в области педиатрии · Jelske W van der BurgOlaf Dammann
30 ноября 2011 г. · Progress in Neuro-psychopharmacology & Biological Psychiatry · Urs Meyer
30 апреля 2011 г. · National Journal of Development Нейронаука: Официальный журнал Международного общества нейробиологии развития·Сандра РизДэвид Уокер
6 ноября 2007 г.·Международный журнал развития неврологии: Официальный журнал Международного общества нейробиологии развития·Сандра РизДэвид Уокер
25 января 2011 г.· Journal of Neuroscience Research·Joseph M AntonyFreda D Miller
4 июля 2012·Нейробиология развития·Elaine Y Hsiao, Paul H Patterson
2 апреля 2014·Glia·Stephen A Back, Paul A Rosenberg
25 августа 2015· Frontiers in Cellular Neuroscience·Mingju CaoMartin G Frasch
25 августа 2015 г.·Frontiers in Cellular Neuroscience·Silke SmoldersBert Brône
1 апреля 7, 2013 · Прогресс в нейроскофармакологии и биологической психиатрии · Ганезанский веккатасубраманский, Monojit debnath
апрель 29, 2014 · Репродуктивная токсикология · Ana Baburamanicanina Marlard
16 мая 2009 г. · Нейробиология и биобехавиоральные отзывы · Урс Мейерс Хоссеин Fatemi
26 декабря 2015 г. · Репродуктивные науки · Alex XuBryan S Richardson
16 июля 2011 г. · Неврология развития · Melinda CzehAngela M Kaindl
21 февраля 2019 г. · Physiological Reports · Susan YS FengAdrian American M Walker
0 26 Dec, Journal 26, Journal физиологии.Регуляторная, интегративная и сравнительная физиология · Сьюзен И. С. Фенг Адриан М. Уокер
15 января 2010 г. · Американский журнал физиологии. Regulatory, Integrative and Comparative Physiology. Susan YS FengAdrian M Walker
5 ноября 2011 г. Journal of Applied Physiology · Lydia L ShookAndrea G Edlow
27 июля 2017 г.· Frontiers in Cellular Neuroscience·Amin MottahedinCarina Mallard
25 октября 2020 г.·Nature Neuroscience·Gun-Hoo ParkSangmi Chung
15 декабря 2020·A Bardia SurgeryJournal of CoonsAlan W Flake
13 марта 2021 г. · Journal of Neuroinflammation · Garima SinghTate Gisslen
16 октября 2018 г. · Biological Psychiatry · Serena B Gumusoglu, Hanna E Stevens
18 июня 2021 г. · 0C0FL Hentoque Communications
11 октября 2019 г. · Журнал торакальной и сердечно-сосудистой хирургии · Кендалл М. Лоуренс Дж. Уильям Гейнор
NFAT5 управляет отложением фибрина и макрофагами, вызванными гиперосмотическим стрессом возрастная инфильтрация через PAI-1 в эндотелии
Научно-исследовательская работа Том 13, Выпуск 3 стр. 3661—3679
Пингпинг Ма
1,3, *, , Гуан Ли 2, *, , Сяоруй Цзян 1, , Синькун Шен 3, , Хун Ли 4, , Ли Ян 1, , Ванцянь Лю 1, ,- 1 Ключевая лаборатория биореологических наук и технологий, Министерство образования, Колледж биоинженерии, Чунцинский университет, Чунцин 400044, Китай
- 2 Онкологическая больница при Чунцинском университете, отделение лучевой терапии, Чунцинский университет, Чунцин 400044, Китай
- 3 Стоматологическая школа и больница Медицинского университета Вэньчжоу, Вэньчжоу 325027, Китай
- 4 Отделение акушерства и гинекологии, больница Синьцяо, Военно-медицинский университет, Чунцин 400037, Китай
* Равный взнос
Получено: 1 июня 2020 г. Принято: 31 октября 2020 г. Опубликовано: 19 декабря 2020 г.
https://дои.org/10.18632/aging.202330Как цитировать
Процитировать эту статью
Как цитировать
Ма П, Ли Г, Цзян Х, Шэнь Х, Ли Х, Ян Л, Лю В, . NFAT5 направляет индуцированное гиперосмотическим стрессом отложение фибрина и инфильтрацию макрофагов через PAI-1 в эндотелии. Старение (Олбани, штат Нью-Йорк). 2021; 13:3661-3679. https://doi.org/10.18632/aging.202330
Скопировать или скачать ссылка:
Выберите нужный формат из списка ниже
Провайдер: ТЫ — ДЖУР AU — Ма, Пингпин AU — Ли, Гуан AU — Цзян, Сяоруй AU — Шэнь, Синькунь AU — Ли, Хонг AU — Ян, Ли AU — Лю, Ванцянь TI — NFAT5 направляет индуцированное гиперосмотическим стрессом отложение фибрина и инфильтрацию макрофагов через PAI-1 в эндотелии. Джо — JA – Старение (Олбани, штат Нью-Йорк) ВЛ — 13 ИС — 3 ПБ — СН — УР — https://doi.орг/10.18632/aging.202330 ДО — 10.18632/стар.202330 СП — 3661 ЭП-3679 ПЯ — АБ — Хотя стресс может значительно способствовать развитию атеросклероза, основные механизмы до сих пор полностью не изучены. Здесь мы успешно показали, что индуцированный высоким содержанием солей ядерный фактор активированных Т-клеток 5 (NFAT5) контролирует эндотелиально-зависимую фибринолитическую активность и экспрессию молекул, связанных с воспалительной адгезией, посредством регуляции ингибитора активатора плазминогена-1 (PAI-1).Мы впервые обнаружили, что диеты с высоким содержанием соли вызывают экспрессию NFAT5 и PAI-1 в эндотелии, что вызывает отложение фибрина и инфильтрацию макрофагов в атеросклеротических артериях мышей ApoE-/-. Сверхэкспрессия NFAT5 увеличивала опосредованную PAI-1 антифибринолитическую активность и активировала гены, связанные с воспалительной адгезией, в эндотелиальных клетках. Нокдаун NFAT5 с помощью siRNA ингибировал экспрессию PAI-1, антифибринолитических и адгезивных молекул. Кроме того, анализ иммунопреципитации хроматина показал, что высокое потребление соли значительно способствует связыванию NFAT5 с промотором PAI-1 (TGGAATTATTT) в эндотелиальных клетках.Наше исследование показало, что NFAT5 обладает большим потенциалом для активации опосредованной PAI-1 фибринолитической дисфункции и адгезии воспалительных клеток, тем самым способствуя атеросклерозу, индуцированному высоким содержанием солей. Скорая помощь —Нажмите, чтобы скопировать эту цитату из текстового поля выше или загрузите цитату:
Авторские права: © 2020 Ma et al. Это статья в открытом доступе, распространяемая на условиях лицензии Creative Commons Attribution License (CC BY 3.0), что разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Аннотация
Хотя стресс может значительно способствовать развитию атеросклероза, лежащие в его основе механизмы до сих пор полностью не изучены. Здесь мы успешно показали, что индуцированный высоким содержанием солей ядерный фактор активированных Т-клеток 5 (NFAT5) контролирует эндотелиально-зависимую фибринолитическую активность и экспрессию молекул, связанных с воспалительной адгезией, посредством регуляции ингибитора активатора плазминогена-1 (PAI-1).Сначала мы наблюдали, что диеты с высоким содержанием соли вызывают экспрессию NFAT5 и PAI-1 в эндотелии, что вызывает отложение фибрина и инфильтрацию макрофагов в атеросклеротических артериях мышей ApoE -/-. Сверхэкспрессия NFAT5 увеличивала опосредованную PAI-1 антифибринолитическую активность и активировала гены, связанные с воспалительной адгезией, в эндотелиальных клетках. Нокдаун NFAT5 с помощью siRNA ингибировал экспрессию PAI-1, антифибринолитических и адгезивных молекул. Кроме того, анализ иммунопреципитации хроматина показал, что высокое потребление соли значительно способствует связыванию NFAT5 с промотором PAI-1 (TGGAATTATTT) в эндотелиальных клетках.Наше исследование показало, что NFAT5 обладает большим потенциалом для активации опосредованной PAI-1 фибринолитической дисфункции и адгезии воспалительных клеток, тем самым способствуя атеросклерозу, индуцированному высоким содержанием солей.
Сокращения
АС: атеросклероз; ЭК: эндотелиальные клетки; HS: высокое содержание соли; PAI-1: ингибитор активатора плазминогена-1; PLAU: активатор урокиназы плазминогена; PLAT: активатор тканевого плазминогена; NFAT5: ядерный фактор активированных Т-клеток 5; vWF: фактор фон Виллебранда; Мыши ApoE -/-: мыши с дефицитом аполипопротеина Е; АА: дуга аорты; ТА: грудная аорта; HUVECs: эндотелиальные клетки пупочной вены человека; ICAM-1: молекула межклеточной адгезии-1; MCP-1: хемоаттрактантный белок-1 моноцитов; VCAM-1: молекула адгезии сосудистых клеток-1; PLG: плазминоген; ЧИП: иммунопреципитация хроматина; ORE: элемент осмотического отклика; β-ECGF: фактор роста β-эндотелиальных клеток.
МиР-146a регулирует воспалительную инфильтрацию макрофагами при полимиозите/дерматомиозите путем нацеливания на TRAF6 и воздействия на путь IL-17/ICAM-1 — FullText — Cellular Physiology and Biochemistry 2016, Vol. 40, № 3-4
Аннотация
Предыстория/цели: Основная цель этого исследования заключалась в изучении роли миР-146a в индуцировании воспалительной инфильтрации макрофагов при полимиозите/дерматомиозите (PM/DM) посредством нацеливания на фактор 6, ассоциированный с рецептором TNF (TRAF6). , который может дополнительно подавлять путь интерлейкина-17 (IL-17) / межклеточной молекулы адгезии 1 (ICAM-1). Методы: Биопсии были взяты у пациентов с ПМ/ДМ и здоровых добровольцев. Создание модели PM/DM и выделение макрофагов проводили на крысах Sprague Dawley (SD). Модельных крыс и макрофаги обрабатывали анти-IL-17, анти-ICAM-1, миметиками miR-146a, ингибиторами miR-146a и миРНК TRAF6. Экспрессию креатинфосфокиназы в сыворотке (S-CK) оценивали с помощью сэндвич-ферментного иммуноферментного анализа (ELISA) с двойными антителами, а для анализа экспрессии CD163 в образцах мышц проводили иммуногистохимический анализ.Кроме того, мы использовали анализ Transwell для проверки миграции клеток; Для определения экспрессии miR-146a, TRAF6, IL-17 и ICAM-1 проводили RT-PCR и вестерн-блоттинг. Результаты: Уровни S-CK, TRAF6, IL-17 и ICAM-1 были выше у пациентов с ПМ/СД по сравнению со здоровым контролем и снижались после традиционного лечения. Лечение миметиками miR-146a, анти-IL-17 и анти-ICAM-1 снижало экспрессию IL-17 и ICAM-1, тогда как ингибиторы miR-146a оказывали противоположное действие.Эффекты ингибиторов miR-146a подавлялись обработкой миРНК TRAF6. Кроме того, репортерный анализ люциферазы подтвердил связь нацеливания между miR-146a и TRAF6. Выводы: МиР-146a регулирует инфильтрацию воспалительных макрофагов при ПМ/ДМ, воздействуя на TRAF6 и воздействуя на путь IL-17/ICAM-1.
© 2016 Автор(ы) Опубликовано S. Karger AG, Basel
Введение
Полимиозит/дерматомиозит (ПМ/ДМ), который характеризуется меристической слабостью проксимальных мышц и повышением уровня мышечных ферментов, является необычной идиопатической воспалительной миопатией [1].Зарегистрированная заболеваемость составляет от 1 на 250 000 до 1 на 130 000 в год, а распространенность ПМ/ДМ составляет приблизительно 1 на 14 000 согласно предыдущим отчетам [2]. Исследователи начали исследовать основную патологию ПМ/ДМ, связанную с аномальной регуляцией аутоиммунитета. Кроме того, представляется важным сочетание генетических полиморфизмов и факторов окружающей среды [1]. В результате не существует теории, основанной на доказательствах, для определения подходящего лечения ПМ/ДМ, а опыт врачей и эмпирические данные играют ключевую роль в выборе лечения [3].Кроме того, своевременная диагностика ПМ затруднена, поскольку его симптомы очень похожи на симптомы миозита [4]. Таким образом, идентификация миозита при ПМ/ДМ поможет клиницистам в определении ключевых факторов риска, имеющих решающее значение для выбора лечения, что представляет собой прорыв в клинической практике [3].
Интерлейкин-17 (ИЛ-17), продуцируемый Т-хелперными клетками, является провоспалительным цитокином и индуцирует TNFα, IL-lb, IL-6, IL-23 и G-CSF [5]. Поскольку IL-17 также был обнаружен в сыворотке пациентов с различными заболеваниями, его связывают с развитием различных заболеваний человека [6], включая рассеянный склероз [7], системную красную волчанку, отторжение аллотрансплантата и астму [8]. ].Молекула межклеточной адгезии 1 (ICAM-1), также известная как CD54, является членом надсемейства иммуноглобулинов [9] и экспрессируется как в растворимой форме, так и в связанной с мембраной форме на поверхности нескольких типов клеток, включая эндотелиальные клетки и иммунные клетки; кроме того, ICAM-1 индуцируется цитокинами, такими как TNF-α, интерлейкин-1 и бактериальный липополисахарид [10]. ICAM-1 выявляется в сыворотке крови и спинномозговой жидкости при инфекционных, аутоиммунных или пролиферативных заболеваниях [11].Недавние данные свидетельствуют о синергии интерлейкина-17 и IFNγ или TNFα в стимулировании экспрессии ICAM-1 при многих воспалительных заболеваниях [12]. Поэтому мы подозревали значительную роль IL-17-ICAM-1 в развитии полимиозита.
МикроРНК (миРНК) представляют собой семейство малых эндогенных некодирующих РНК и участвуют в регуляции множества биологических процессов, начиная от определения клеточной судьбы и заканчивая сигнальными событиями на посттранскрипционном уровне [13,14]. МиР-146a является членом семейства микроРНК миР-146 и имеет ту же исходную последовательность, что и миР-146b-5p.МиР-146a модулирует как врожденный, так и адаптивный иммунитет [15] путем негативной регуляции фактора 6, ассоциированного с рецептором TNF (TRAF6), и киназы 1, ассоциированной с рецептором IL-1 (IRAKI), двух адапторных белков, оба из которых важны для про- воспалительная сигнализация [16]. Повышающая регуляция miR-146a подавляет экспрессию IRAK1/TRAF6, впоследствии ингибируя передачу сигналов NF-κB и экспрессию нижестоящих белков-мишеней (ICAM-1, VCAM-1, IL-6 и PGE2) [17]. В мононуклеарных клетках периферической крови (РВМС) и синовиальной оболочке у пациентов с ревматоидным артритом miR-146a ассоциирована с экспрессией IL-17 [5].Поэтому мы предположили, что миР-146a связана с путем IL-17/ICAM-1 при полимиозите. Основная цель этого исследования заключалась в изучении роли miR-146a в индукции воспалительной инфильтрации макрофагов при PM/DM путем нацеливания на TRAF6, который может дополнительно активировать путь IL-17/ICAM-1.
Материалы и методы
Клинические образцы
У 20 пациентов (мужчины : женщины = 6:14) среднего возраста 61 год был диагностирован полимиозит (ПМ) или дерматомиозит (ДМ) по критериям Bohan и Peter [18] в Первой дочерней больнице Китайского медицинского университета в период с марта 2014 г. по октябрь 2014 г.Все пациенты получали высокие дозы глюкокортикоидов или других иммунодепрессантов, включая метотрексат, циклофосфамид и азатиоприн. Семь женщин и трое мужчин без клинических или гистопатологических признаков какого-либо заболевания мышц были включены в контрольную группу (средний возраст: 56 лет). Клинические характеристики участников представлены в таблице 1. У каждого пациента были взяты две биопсии: одна была получена до лечения, а другая — через 8 месяцев после лечения глюкокортикоидами.Образцы тканей, собранные в полуоткрытом анализе, затем замораживали при -80°C [19]. Одна клиническая оценка состояла из функционального индекса миозита (FI), который измеряет количество выполненных повторений в различных группах мышц и был представлен в процентах от максимального балла 64. Это исследование было рассмотрено и одобрено институциональным комитетом по этике Первого аффилированного Госпиталь Китайского медицинского университета, согласно Хельсинкской декларации. Перед включением в исследование от пациентов было получено информированное согласие.
Таблица 1
Клинические данные о пациентах и здоровом контроле. М, мужчина; Ф, женщина; нет данных, нет данных; ПМ, полимиозит; СД, дерматомиозит; АНА, антинуклеарное антитело; Ro52, антиген типа А синдрома Шегрена; Ro60, антиген А2 синдрома Шегрена; Mi-2, хромодомен-хеликаза-ДНК-связывающий белок; SRP, частица распознавания сигнала; Пред, преднизолон; метотрексат, метотрексат; АЗА, азатиоприн; s-CK, сывороточная креатинфосфокиназа; FI, функциональный индекс
Животная модель
Сорок самцов крыс Sprague Dawley (SD) (масса: 225-350 г, возраст: 2-3 месяца) были получены из Китайского медицинского университета.Животные модели полимиозита были созданы путем множественных абдоминальных инъекций гомогената скелетных мышц кролика, как описано ранее [20]. Крысы были случайным образом разделены на 4 экспериментальные группы: контрольная группа (лечение плацебо), модельная группа (установленная модель полимиозита), группа анти-IL-17 (крысы с моделью полимиозита, которым ежедневно вводили 35 мкКи моноклонального антитела против IL-17). (мАт) после 14 -го -го дня после операции) и группу анти-ICAM-1 (крысы с моделью полимиозита, которым ежедневно вводили 35 мкКи анти-ICAM-1 mAb после 14 -го -го дня после операции) .Все крысы были проверены и подвергнуты эвтаназии через 4 недели после операции.
Тесты на сывороточную креатинфосфокиназу (S-CK), IL-17 и ICAM-1
Уровни S-CK, IL-17 и ICAM-1 оценивали в образцах крови, извлеченных из каждой группы, путем проведения сэндвич-фермента с двойным антителом сцепленный иммуноферментный анализ (ИФА) (R&D Systems, Миннеаполис, Миннесота, США) в соответствии с инструкцией к набору.
Прилипание клеточной культуры
Перитонеальные макрофаги выделяли из крыс SD.Фосфатно-солевой буфер (PBS), содержащий 3% раствор тиогликолята Брюера, вводили в брюшную полость крыс SD. Через три дня брюшную полость промывали PBS и собирали клетки макрофагов. После процеживания через сито 70 мкм и промывания буфером аммоний-хлорид-калий (ACK) клетки ресуспендировали в среде Roswell Park Memorial Institute (RPMI). После 2-часового периода культивирования при 37°C в инкубаторе с 5% CO 2 клетки макрофагов выделяли с помощью PBS и поддерживали в RPMI.Макрофаги были разделены на 7 групп: контрольная группа (клетки без обработки), группа миметиков miR-146a (клетки, трансфицированные миметиками miR-146a), группа ингибиторов miR-146a (клетки, трансфицированные ингибиторами miR-146a), группа ингибиторов miR-146a + TRAF6 siRNA (клетки, трансфицированные ингибиторами miR-146a и TRAF6 siRNA), группа miR-146a mimics негативный контроль (миметики NC, клетки, трансфицированные miR-146a имитирует отрицательный контроль), ингибиторы miR-146a группа отрицательного контроля (ингибиторы NC, клетки, трансфицированные ингибиторами miR-146a, отрицательный контроль) и группа отрицательного контроля siRNA (NC siRNA, клетки, трансфицированные siRNA без конкретной последовательности).Миметики, ингибиторы, миметики отрицательного контроля, ингибиторы отрицательного контроля и siRNA были приобретены у Santa Cruz Biotechnology (Санта-Круз, Калифорния, США) и трансфецированы в клетки с использованием липофектамина RNAiMAX (Invitrogen, Карлсбад, Калифорния, США) в указанных концентрациях.
Иммуногистохимия
Перед проведением гистологического анализа мышечные ткани обрабатывали 30% раствором сахарозы и 4% параформальдегидом для перфузии. Замороженные ткани разрезали на срезы, на которые затем наносили первичные антитела против CD163 (разведение 1:1000, BD Biosciences, Сан-Хосе, Калифорния, США).Затем образцы инкубировали с конъюгированными с пероксидазой хрена (HRP-конъюгированными) козьими антикроличьими IgG (1:800, Zhongshan Biology Company, Пекин, Китай), вторичными антителами. После визуализации с помощью диаминобензидина (DAB, Sigma-Aldrich, Сент-Луис, Миссури, США) образцы гидратировали, промывали и анализировали под микроскопом. В группе отрицательного контроля клетки обрабатывали неспецифическими IgG (1:800, Zhongshan Biology Company, Пекин, Китай) для замены первичных антител.
Анализ активности люциферазы
Последовательность 3′-нетранслируемой области (UTR) TRAF6, содержащей сайт связывания миР-146a, амплифицировали с помощью полимеразной цепной реакции (ПЦР) и клонировали в нижний сайт векторов люциферазы pGL3 (Promega, Madison , Висконсин, США).Мутированный сайт связывания получали с использованием системы сайт-направленного мутагенеза GeneTailor (Invitrogen, Карлсбад, Калифорния, США), а затем мутантную 3′-UTR также клонировали в векторы pGL3-люциферазы (соответствующие последовательности праймеров перечислены в таблице 2). Макрофаги содержали в 96-луночных планшетах. Репортерные векторы, которые несут 3′-UTR TRAF6 или мутантную 3′-UTR TRAF6, или контрольные последовательности котрансфицировали 200 нг репортера pGL3-control люциферазы и 10 нг векторов pRL-TK (репортеры внутреннего контроля).В анализе использовали репортерную систему Dual-Luciferase Assay System (Promega, Мэдисон, Висконсин, США) с тремя повторными экспериментами в соответствии с инструкциями набора. Клетки собирали через 48 ч и определяли относительную активность люциферазы.
Таблица 2
Последовательности праймеров для экспериментов с репортером люциферазы. UTR: нетранслируемая область
Анализ Transwell
Клетки (2 x 10 5 ) голодали в течение 24 часов, а затем переносились в 24-луночный Transwell с мембранной вставкой 8 мкм (Corning, Corning, NY, USA) после ресуспендирование в бессывороточной среде F12.Среду F12, содержащую 1% гормондефицитной сыворотки и 100 нг/мл МСР-1, использовали в качестве хемоаттрактанта и помещали в нижнюю камеру. После инкубации в течение 72 ч клетки, проникшие через мембрану, фиксировали 95%-ным этанолом и окрашивали кристаллическим фиолетовым. Затем инвазированные клетки наблюдали и подсчитывали под микроскопом.
Выделение РНК и ОТ-ПЦР
Тотальную РНК из тканей и клеток выделяли с использованием реагента TRIzol (Invitrogen, Карлсбад, Калифорния, США) в соответствии с инструкциями производителя.Набор ReverTra Ace qPCR RT Kit (Toyobo Co., Ltd, Кита-ку, Осака, Япония) использовали для транскрипции тотальных РНК в кДНК, а RT-PCR проводили с использованием THUNDERBIRD SYBR® qPCR Mix (Toyobo Co., Ltd, Kita). -ku, Осака, Япония) с помощью прибора CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Последовательности праймеров приведены в таблице 3. Уровни экспрессии генов-мишеней нормализовали по уровню экспрессии глицеральдегид-3-фосфатреальной дегидрогеназы (GAPDH), и для расчетов использовали метод 2 -ΔΔCt .
Таблица 3
Последовательности праймеров ОТ-ПЦР для GAPDH и миР-146a. GAPDH: фосфоглицеральдегиддегидрогеназа, RT-PCR: почечная полимеразная цепная реакция
Вестерн-блоттинг
Ткани и клетки собирали и лизировали буфером RIPA. Суммарные белки разделяли и подсчитывали по методу Брэдфорда [21]. Затем общий белок денатурировали в кипящей воде и переносили на мембрану из поливинилиденфторида (PVDF) после электрофореза в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE).Мембрану блокировали в TBST с 5% обезжиренным молоком в течение 1 ч и обрабатывали первичными антителами против TRAF6, IL-17, ICAM-1 (1:800, 1:500 и 1:800 соответственно; Zhongshan Biology Company, Пекин). , Китай) при 4°C в течение ночи. После промывки мембрану инкубировали со вторичными антителами (конъюгированные с пероксидазой хрена козьи антикроличьи антитела, разведение 1:2000; Zhongshan Biology Company, Пекин, Китай). Затем образцы были визуализированы с усиленной хемилюминесценцией и количественно оценены с помощью Lab Works 4.5 с GAPDH в качестве эндогенного контроля.
Статистический анализ
Все статистические анализы проводились с помощью программного обеспечения SPSS 18.0 (версия X; IBM, Армонк, штат Нью-Йорк, США). Данные представлены как среднее ± стандартное отклонение, рассчитанное по крайней мере из трех повторных экспериментов. Двусторонний критерий Стьюдента t или однофакторный дисперсионный анализ (ANOVA) использовали для анализа межгрупповых сравнений среди более чем двух групп; P < 0,05 считалось статистически значимым.
Результаты
Клинические оценки, гистопатология мышц и эффекты традиционной гормональной терапии
Положительное окрашивание CD163 было обнаружено среди мононуклеарных воспалительных клеток, особенно макрофагов. Экспрессия CD 163 не наблюдалась в образцах мышц здоровых контролей (рис. 1С) как до лечения (рис. 1А), так и после лечения (рис. 1В). Окрашенная область, представляющая экспрессию CD163, продуцируемую макрофагами, уменьшилась после гормональной обработки.
Рис.1
Иммуногистохимическое окрашивание на CD163 (окрашено коричневым цветом) в образцах мышц пациентов до лечения (A), после лечения (B) и контрольной группы здоровых людей (C).
По сравнению со здоровым контролем мышечная активность до лечения в группе PM/DM была намного ниже, но при обычном гормональном лечении наблюдалось заметное улучшение. Точно так же уровень s-CK повышался у пациентов до лечения по сравнению со здоровыми людьми из контрольной группы и снижался после гормонального лечения (рис.2А, Таблица 1).
Рис. 2
Уровни экспрессии s-CK (A), miR-146a (B), TRAF6 (C), IL-17 (D) и ICAM-1 (E) у пациентов до лечения, после лечения и здоровый контроль. Данные представлены как среднее значение ± стандартное отклонение. ** P < 0,01 по сравнению с пациентами в группе до лечения, # P < 0,05, ## P < 0,01 по сравнению с пациентами в группе после лечения. S-CK: сывороточная креатинфосфокиназа.
Экспрессия miR-146a, TRAF6, IL-17 и ICAM-1 в образцах мышц пациентов и здорового контроля
По сравнению с контрольной группой экспрессия miR-146a была значительно снижена у пациентов как до лечения, так и после терапии, а после традиционной гормональной терапии наблюдалось резкое увеличение экспрессии miR-146a (все P < 0.05, рис. 2Б). Экспрессия TRAF6, IL-17 и ICAM-1 продемонстрировала противоположную тенденцию экспрессии по сравнению с miR-146a, т.е. пациенты в контрольной группе демонстрировали самые низкие уровни, тогда как пациенты в группе пациентов до лечения имели самые высокие уровни TRAF6, IL -17 и ICAM-1, и были значительные различия между группой пациентов до лечения, группой пациентов после лечения и контрольной группой (все P < 0,05, рис. 2C-E).
Экспрессия CD-163, s-CK, miR-146a, TRAF6, IL-17 и ICAM-1 в образцах мышц крыс с моделью полимиозита
Образцы мышц из контрольной, модельной, анти-IL-17 и Группы анти-ICAM-1 окрашивали анти-CD163, чтобы выявить инфильтрацию воспалительных макрофагов.CD163 почти не наблюдался в контрольной группе (рис. 3А), но продемонстрировал заметное увеличение в модельной группе (рис. 3В). Лечение анти-IL-17 или анти-ICAM-1 приводило к выраженному подавлению экспрессии CD163 (фиг. 3C-D).
Рис. 3
Иммуногистохимическое окрашивание CD 163 (окраска коричневым) у модельных крыс из контрольной (А), модельной (В), анти-IL-17 (С) и анти-ICAM-1 (D) групп .
Как показано на рис. 4A, контрольная группа продемонстрировала самые низкие уровни экспрессии s-CK, тогда как модельная группа показала значительно более высокую экспрессию s-CK, чем контрольная группа ( P < 0.05). Экспрессия S-CK в группах анти-IL-17 и анти-ICAM-1 была значительно ниже, чем в экспериментальной группе, но все же выше, чем в контрольной группе ( P < 0,05), тогда как между группа анти-IL-17 и группа анти-ICAM-1 по экспрессии s-CK ( P > 0,05).
Рис. 4
Экспрессия s-CK, miR-146a, TRAF6, IL-17 и ICAM-1 у модельных крыс. (A) Уровни s-CK у крыс, получавших различное лечение (т.е., контроль, модель, анти-IL-17 и анти-ICAM-1). (B) Количественные данные об уровнях экспрессии миР-146a у модельных крыс. (C) Количественные данные об уровнях экспрессии мРНК TRAF6 у модельных крыс. (D-E) Количественные уровни белка IL-17 (D) и ICAM-1 (E) у модельных крыс. (F) Вестерн-блот анализ IL-17 и ICAM-1 у модельных крыс с GAPDH в качестве внутреннего контроля. Данные представлены как среднее значение ± стандартное отклонение. ** P < 0,01 по сравнению с контрольной группой, # P < 0.05, ## P < 0,01 по сравнению с модельной группой, ° P < 0,05, ºº P < 0,01 по сравнению с группой анти-IL-17.
Как показано на рис. 4B-F, по сравнению с контрольной группой, модельная группа продемонстрировала значительно более высокую экспрессию TRAF6, IL-17 и ICAM-1, но более низкую экспрессию miR-146a (все P < 0,05 ). Лечение анти-IL-17 значительно снижало экспрессию IL-17 и ICAM-1 ( P < 0.05), но мало влиял на экспрессию miR-146a ( P > 0,05). Кроме того, лечение анти-ICAM-1 сильно снижало экспрессию ICAM-1 ( P < 0,05), но не влияло на экспрессию miR-146a и IL-17 ( P > 0,05).
МиР-146а подавляет экспрессию TRAF6 путем связывания с 3′-НТО
Один высококонсервативный сайт связывания миР-146а был предсказан в 3′-НТО TRAF6 с помощью программного обеспечения TargetScan (рис. 5А).Анализ активности люциферазы использовали для оценки прямого взаимодействия между miR-146a и TRAF6. Как показано на фиг. 5В, по сравнению с группой NC, только трансфекция 3′-UTR TRAF6 дикого типа показала значительное снижение относительной активности люциферазы ( P < 0,05), тогда как в клетках, трансфицированных мутацией TRAF6-3, различий не наблюдалось. UTR и miR-146a из соответствующей контрольной группы ( P > 0,05).
Рис. 5
Предсказание сайта связывания и результаты анализа люциферазы.(A) Предполагаемые цели, предсказанные TargetScan. (B) Относительная активность люциферазы в результате связывания 3’UTR TRAF6 и miR-146a в макрофагах через 48 часов после трансфекции. * P < 0,05 по сравнению с группой NC. WT: TRAF6-3' UTR дикого типа; Mut: TRAF6 mut-3'UTR; НК: отрицательный контроль.
MiR-146a/TRAF6 регулирует миграцию макрофагов
Количество мигрировавших клеток в анализе Transwell существенно уменьшилось после трансфекции миметиками miR-146a по сравнению с группой миметиков NC ( P < 0.05, рис. 6). Обработка ингибиторами miR-146a приводила к заметному увеличению количества мигрирующих клеток по сравнению с обработкой ингибиторами NC, тогда как этому эффекту противодействовала трансфекция миРНК TRAF6 ( P < 0,05, рис. 6).
Рис. 6
МиР-146a активирует миграцию макрофагов. (A) Количество клеток, которые проникли через мембрану вставки в контроле, миметиках NC, ингибиторах NC, миРНК NC, миметиках миР-146а, ингибиторах миР-146а, ингибиторах миР-146а + группах миРНК TRAF6.(B) Количество мигрирующих клеток. Данные представлены как среднее ± стандартное отклонение для трех независимых экспериментов. ** P < 0,01 по сравнению с группой миметиков NC, ## P < 0,01 по сравнению с группой ингибиторов NC, °° P < 0,01 по сравнению с группой миметиков miR-146a.
MiR-146a/TRAF6 регулируют экспрессию IL-17 и ICAM-1
По сравнению с контрольной группой группа миметиков miR-146a продемонстрировала значительно более высокие уровни экспрессии miR-146a.В то время как группа ингибиторов miR-146a демонстрировала значительно более низкие уровни экспрессии miR-146a (все P < 0,05), не было существенной разницы между ингибиторами miR-146a и группой ингибиторов miR-146a + TRAF6 siRNA ( P > 0,05, рис. 7А).
Рис. 7
МиР-146a регулирует экспрессию IL-17 и ICAM-1. (A) Количественные данные об уровнях экспрессии миР-146a в клетках макрофагов. (B-D) Количественные уровни белка TRAF6 (B), IL-17 (C) и ICAM-1 (D) в клетках макрофагов.E: Вестерн-блот анализ TRAF6, IL-17 и ICAM-1 в клетках макрофагов с GAPDH в качестве внутреннего контроля. Данные представлены как среднее ± стандартное отклонение для трех независимых экспериментов. * P < 0,05, ** P < 0,01 по сравнению с группой имитаторов NC, ## P < 0,01 по сравнению с группой ингибиторов NC, °° P 0,01 группа.
Что касается экспрессии TRAF6 в трансфицированных клетках, группа миметиков miR-146a продемонстрировала значительно более низкую экспрессию, тогда как группа ингибиторов miR-146a продемонстрировала значительно более высокую экспрессию по сравнению с группами ингибиторов NC и ингибиторов miR-146a + TRAF6 siRNA ( все P < 0.05).
Как показано на рис. 7C, D, E, по сравнению с группой миметиков NC, трансфекция миметиками miR-146a значительно снижает экспрессию IL-17 и ICAM-1, тогда как трансфекция ингибиторами miR-146a резко повышал экспрессию этих молекул ( P < 0,05). Экспрессия IL-17 и ICAM-1 в группе ингибиторов miR-146a + миРНК TRAF6 была значительно ниже, чем в группе ингибиторов miR-146a, но выше, чем в группе миметиков miR-146a ( P < 0.05).
Обсуждение
Как две идиопатические воспалительные миопатии (ИВМ), полимиозит и дерматомиозит (ПМ/ДМ) характеризуются хронической мышечной слабостью и воспалением в мышечной ткани, сопровождающимся повышением уровня сывороточной креатининкиназы (s-CK), что приводит к инвалидности и другим осложнениям [22]. До сих пор мы мало знали о конкретном патогенезе ПМ/ДМ, хотя несколько исследований продемонстрировали важную роль клеточных воспалительных реакций в прогрессировании ПМ/ДМ [23,24].Т- и В-клетки широко распространены в скелетных мышцах больных ПМ/СД [25]. В дополнение к иммунным механизмам неиммунные механизмы также рассматриваются как факторы, способствующие патогенезу ПМ/ДМ, включая стресс эндоплазматического ретикулума и аутофагию при гибели и дисфункции клеток скелетных мышц при миозите [26]. Однако конкретный вклад этих путей в процесс PM/DM до конца не изучен. Необходимо провести большое количество исследований, прежде чем мы сможем определить механизмы, лежащие в основе ПМ/ДМ.
В этом исследовании, проведенном на пациентах с ПМ/ДМ, мы наблюдали значительное повышение уровней IL-17 и ICAM-1 по сравнению со здоровой контрольной группой. Патогенетическое развитие сопровождалось усилением микрофагальной инфильтрации, купированием этого воспалительного состояния способствовала традиционная гормональная терапия. Во многих исследованиях изучалась взаимосвязь между IL-17, ICAM-1 и PM [27,28,29,30]. Предыдущие исследования выявили сильное влияние ИЛ-17 на патогенез ряда других аутоиммунных заболеваний, в том числе псориаза [31], ревматоидного артрита [32] и рассеянного склероза [33], что приводит к развитию направленных на ИЛ-17 моноклональных терапии антителами.Среди пациентов с ПМ/СД отмечаются более высокие уровни IL-17 [34,35]. Кроме того, в мышечной ткани IL-17 действует в сочетании с другими провоспалительными цитокинами, продуцируемыми моноцитами и врожденными иммунными реакциями, усиливая иммунные реакции, потенциально приводящие к разрушению мышц [36]. Однако использование ICAM-1 связано с образованием микрососудов и может способствовать активации эндотелиальных клеток (ЭК) [37]. В последние годы в ряде исследований выявлена важная роль сосудистой системы в патогенезе ПМ/СД [38,39,40].Изменения в микрососудах могут влиять на местное кровообращение в мышцах, что приводит к пуллюляции симптомов гипоксии. Постоянно региональная гипоксия в мышечной ткани приводит к миастении и мышечной усталости, что является признаком ранней стадии ПМ/СД [41]. Таким образом, и ICAM-1, и IL-17 способствуют ухудшению ПМ/ДМ, и их роль в этом процессе заслуживает дальнейшего внимания.
Напротив, как у пациентов, так и у животных моделей уровни экспрессии miR-146a были намного ниже, чем у здоровых субъектов или в контрольной группе, соответственно.Кроме того, когда имитаторы или ингибиторы miR-146a экспериментально применялись для воздействия на уровни экспрессии miR-146a, мы обнаружили влияние на уровни IL-17 и ICAM-1, а также на миграционную способность макрофагов. Все эти результаты подтвердили нашу гипотезу о том, что миР-146a регулирует экспрессию IL-17 и ICAM-1 у пациентов с ПМ/СД. Предыдущее исследование признало важность miR-146a при PM/DM [42]. Хотя было проведено несколько исследований по изучению специфических регуляторных механизмов, аномальная экспрессия miR-146a была подтверждена исследователями [43].Точно так же наше исследование раскрыло возможный механизм, лежащий в основе регуляции miR-146a экспрессии IL-17 и ICAM-1 при PM/DM.
Мы были удивлены, обнаружив положительное влияние ингибиторов miR-146a на экспрессию IL-17 и ICAM-1, а также миграцию макрофагов, которой противодействовала трансфекция миРНК TRAF6, что согласуется с нашим предположением, что TRAF6 участвует в регуляции миР-146a в ПМ/ДМ. Сообщалось, что MiR-146a воздействует на TRAF6, чтобы изменить течение сахарного диабета 2 типа (T2DM) [44], рака предстательной железы [45] и артрита [46].Являясь одной из наиболее важных сигнальных молекул в клетках, TRAF6 действует непосредственно с рецепторами киназы-1, ассоциированной с рецептором интерлейкина-1 (IRAK), и рецепторами ядерного фактора каппа-В (NF-κB), активируя каналы передачи сигнала [47]. Сигнальный путь NF-κB является важным путем регуляции экспрессии иммунорегуляторных и провоспалительных медиаторов [48], а врожденный иммунный и воспалительный ответы играют решающую роль в патогенезе ПМ/СД [45]. Таким образом, мы полагаем, что miR-146a нацелена на TRAF6 для регуляции патогенеза PM/DM, наблюдение, о котором не сообщалось ни в одном другом исследовании.Кроме того, miR-146a нацелена на TRAF6 и влияет на выработку IL-17 у пациентов с псориазом [49]. Экспрессия ICAM-1 была связана с этим сигнальным путем при некоторых хронических воспалительных заболеваниях, таких как диабетическая сетчатка [50]. Наше исследование продемонстрировало влияние оси miR-146a/TRAF6 на экспрессию IL-17 и ICAM-1, которые затем влияют на патогенез ПМ/СД.
Как уже упоминалось, наш интерес вызвал сигнальный путь NF-κB, и его роль в развитии ПМ/ДМ требует дальнейшего изучения.Мы намеревались провести наши дальнейшие исследования для изучения возможного взаимодействия между осью miR-146a/TRAF6 и сигнальным путем.
Таким образом, это исследование впервые продемонстрировало способность TRAF6, нацеленного на миР-146a, регулировать экспрессию IL-17 и ICAM-1. Это открытие будет способствовать нашему пониманию патогенеза развития ПМ/ДМ и облегчит диагностику. Необходимы дальнейшие исследования для выяснения основного механизма и других регуляторных факторов, а также роли сигнальных белков NF-κB и белков-мишеней.
Заявление о раскрытии информации
Авторы заявляют об отсутствии коммерческих или финансовых конфликтов интересов.
Каталожные номера
- Маммен А.Л.: Дерматомиозит и полимиозит: клиническая картина, аутоантитела и патогенез.Ann NY Acad Sci 2010;1184:134-153.
- Финдли А.Р., Гоял Н.А., Мозаффар Т.: Обзор полимиозита и дерматомиозита. Мышечный нерв 2015; 51: 638-656.
- Dalakas MC: Иммунотерапия миозита: вопросы, проблемы и перспективы на будущее.Nat Rev Rheumatol 2010; 6: 129-137.
- Milisenda JC, Selva-O’Callaghan A, Grau JM: диагностика и классификация полимиозита. J Аутоиммун 2014;48-49:118-121.
- Ниимото Т., Накаса Т., Исикава М., Окухара А., Изуми Б., Дейе М., Судзуки О., Адачи Н., Очи М.: МикроРНК-146а экспрессируется в Т-клетках, продуцирующих интерлейкин-17, у пациентов с ревматоидным артритом.BMC Расстройство опорно-двигательного аппарата 2010; 11:209.
- Накаэ С., Комияма Ю., Намбу А., Судо К., Ивасе М., Хомма И., Секикава К., Асано М., Ивакура И.: У мышей с дефицитом ИЛ-17 нарушена сенсибилизация антиген-специфических Т-клеток, что вызывает подавление аллергических клеточных и гуморальных реакций. ответы. Иммунитет 2002;17:375-387.
- Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H: Экспрессия мРНК интерлейкина-17 в мононуклеарных клетках крови и спинномозговой жидкости увеличивается при рассеянном склерозе. Мульт Склер 1999; 5:101-104.
- Wong CK, Ho CY, Li EK, Lam CW: Повышение концентрации провоспалительных цитокинов (IL-18, IL-17, IL-12) и цитокинов Th3 (IL-4) у пациентов с системной красной волчанкой.Волчанка 2000;9:589-593.
- Холланд Дж., Оуэнс Т.: Передача сигналов через молекулу межклеточной адгезии 1 (ICAM-1) в линии В-клеточной лимфомы. Активация тирозинкиназы Lyn и митоген-активируемого протеинкиназного пути. J Biol Chem 1997; 272:9108-9112.
- Othumpangat S, Noti JD, McMillen CM, Beezhold DH: ICAM-1 регулирует выживаемость вируса гриппа в эпителиальных клетках легких на ранних стадиях инфекции.Вирусология 2016;487:85-94.
- Pietruczuk M, Pietruczuk A, Pancewicz S, Hermanowska-Szpakowicz T: [ICAM-1: структура, биологическая роль и клиническое значение]. Пол Меркур Лекарски 2004; 17: 507-511.
- Габр М.А., Цзин Л., Хелблинг А.Р., Синклер С.М., Аллен К.Д., Шамджи М.Ф., Ричардсон В.Дж., Фитч Р.Д., Сеттон Л.А., Чен Дж.: Интерлейкин-17 взаимодействует с IFNgamma или TNFalpha, способствуя высвобождению медиатора воспаления и молекулы межклеточной адгезии-1 ( Экспрессия ICAM-1) в клетках межпозвонкового диска человека.J Orthop Res 2011; 29: 1-7.
- Айзенберг И., Эран А., Нишино И., Моджио М., Ламперти С., Амато А.А., Лидов Х.Г., Канг П.Б., Норт К.Н., Митрани-Розенбаум С., Фланиган К.М., Нили Л.А., Уитни Д., Беггс А.Х., Кохане И.С., Кункель Л.М.: Отличительные паттерны экспрессии микроРНК при первичных мышечных заболеваниях.Proc Natl Acad Sci USA 2007;104:17016-17021.
- Амброс В.: Функции животных микроРНК. Природа 2004;431:350-355.
- Лофгрен С.Э., Фростегард Дж., Трюдссон Л., Понс-Эстель Б.А., Д’Альфонсо С., Витте Т., Лауверис Б.Р., Эндреффи Э., Ковач Л., Васконселос С., Мартинс да Сува Б., Козырев С.В., Аларкон-Рикельме М.Е.: Генетическая ассоциация miRNA-146a при системной красной волчанке у европейцев за счет снижения экспрессии гена.Genes Immun 2012; 13:268-274.
- Болдин М.Р., Таганов К.Д., Рао Д.С., Ян Л., Чжао Дж.Л., Калвани М., Гарсия-Флорес Й., Луонг М., Девреканли А., Сюй Дж., Сун Г., Тай Дж., Линсли П.С., Балтимор Д.: miR-146a является важным тормозят аутоиммунную миелопролиферацию и рак у мышей. J Exp Med 2011; 208:1189-1201.
- Yang CR, Shih KS, Liou JR, Wu YW, Hsieh IN, Lee HY, Lin TC, Wang JH: Денбинобин активирует экспрессию miR-146a и ослабляет индуцированную IL-lbeta активацию экспрессии ICAM-1 и VCAM-1 в фибробластах при остеоартрите -подобные синовиоциты. J Mol Med (Берл) 2014; 92:1147-1158.
- Троянов Ю., Таргоф И.Н., Пайетт М.Р., Рейно Дж.П., Шартье С., Гуле Дж.Р., Бурр-Тессье Дж., Рич Э., Гродзицкий Т., Фрицлер М.Дж., Джоял Ф., Кениг М., Сенекал Дж.Л.: Новое определение дерматомиозита: описание новых диагностических критериев которые отличают чистый дерматомиозит от перекрывающегося миозита с признаками дерматомиозита.Медицина (Балтимор) 2014; 93: 318-332.
- Хенрикссон К.Г.: «Полуоткрытая» техника биопсии мышц. Простая амбулаторная процедура. Acta Neurol Scand 1979; 59:317-323.
- Хань Л.Н., Ли Т.Л., Дин Г.Л., Лю Дж.В., Дин Ю., Чжан Ю.Дж.: [Создание экспериментальной модели аутоиммунного миокардита, индуцированного человеческим сердечным белком С, у крыс].Чжунхуа Синь Сюэ Гуань Бин За Чжи 2012; 40: 690-696.
- Крюгер, штат Нью-Джерси: Метод Брэдфорда для количественного определения белка. Методы Мол Биол 1994;32:9-15.
- Pandya JM, Loell I, Hossain MS, Zong M, Alexanderson H, Raghavan S, Lundberg IE, Malmstrom V: Эффекты обычного иммуносупрессивного лечения на CD244+ (CD28null) и F0XP3+ T-клетки в воспаленной мышце у пациентов с полимиозитом и дерматомиозитом.Артрит Res Ther 2015;18:80.
- Нотарникола А., Лападула Г., Натуцци Д., Лундберг И.Е., Янноне Ф.: Корреляция между уровнями ИЛ-15 и ИЛ-17 в сыворотке у пациентов с идиопатическими воспалительными миопатиями. Scand J Rheumatol 2015;44:224-228.
- Salomonsson S, Lundberg IE: Цитокины при идиопатических воспалительных миопатиях.Аутоиммунитет 2006;39:177-190.
- Rayavarapu S, Coley W, Nagaraju K: Обновление патогенных механизмов воспалительных миопатий. Curr Opin Rheumatol 2011;23:579-584.
- Henriques-Pons A, Nagaraju K: Неиммунные механизмы повреждения мышц при миозите: роль стрессовой реакции эндоплазматического ретикулума и аутофагии в патогенезе заболевания.Curr Opin Rheumatol 2009;21:581-587.
- Szodoray P, Alex R, Knowlton N, Centola M, Dozmorov I, Csipo I, Nagy AT, Constantin T, Ponyi A, Nakken B, Danko K: идиопатические воспалительные миопатии, на которые указывают характерные периферические цитокины, хемокины и члены семейства TNF B фактор активации клеток и лиганд, индуцирующий пролиферацию.Ревматология (Оксфорд) 2010;49:1867-1877.
- Tournadre A, Porcherot M, Cherin R Marie I, Hachulla E, Miossec P: Баланс Thl и Thl7 при воспалительных миопатиях: взаимодействие с дендритными клетками и возможная связь с реакцией на высокие дозы иммуноглобулинов. Цитокин 2009;46:297-301.
- Wang M, Jia JP, Da YW: [Исследование сосудистого фактора в патогенезе полимиозита]. Чжунхуа Нэй Кэ За Чжи 2005; 44: 115-117.
- Саллум А.М., Кисс М.Х., Сува К.А., Вакамацу А., Вианна М.А., Сачетти С., Мари С.К.: Разница в экспрессии молекул адгезии (ICAM-1 и VCAM-1) при ювенильном и взрослом дерматомиозите, полимиозите и миозите с включениями.Аутоиммун Рев. 2006; 5:93-100.
- Baeten DL, Kuchroo VK: Как цитокиновые сети вызывают воспаление: интерлейкин-17 и рассказ о двух аутоиммунных заболеваниях. Nat Med 2013;19:824-825.
- Kenna TJ, Brown MA: Роль тучных клеток, секретирующих IL-17, в воспалительных заболеваниях суставов.Nat Rev Rheumatol 2013; 9: 375-379.
- Miossec P: Интерлейкин-17 наконец-то в моде: через десять лет после его описания был идентифицирован его клеточный источник. Arthritis Rheum 2007;56:2111-2115.
- Мельник П., Хвалинска-Садовска Х., Весик-Шевчик Э., Маслински В., Олесинска М. Концентрация интерлейкина 15, рецептора интерлейкина 2 и рецептора ФНО в сыворотке крови у пациентов с полимиозитом и дерматомиозитом: корреляция с активностью заболевания.Rheumatol Int 2012;32:639-643.
- Зонг М., Лоэлл И., Линдроос Э., Надер Г.А., Александрсон Х., Халленгрен К.С., Борг К., Арнардоттир С., Маклиннес И.Б., Лундберг И.Е.: Влияние иммуносупрессивного лечения на экспрессию интерлейкина-15 и альфа-рецептора интерлейкина-15 в мышечной ткани пациентов при полимиозите или дерматомиозите.Энн Реум Дис 2012; 71: 1055-1063.
- Tournadre A, Miossec P: Интерлейкин-17 при воспалительных миопатиях. Curr Rheumatol Rep 2012;14:252-256.
- Барбассо Хелмерс С., Энглунд П., Энгстром М., Алин Э., Фати М., Янчаускиен С., Хеймбургер М., Роннелид И., Лундберг И.Е.: Сыворотки анти-Jo-1-положительных пациентов с полимиозитом и интерстициальным заболеванием легких индуцируют экспрессию молекулы межклеточной адгезии 1 в эндотелиальных клетках легких человека.Arthritis Rheum 2009;60:2524-2530.
- Figarella-Branger D, Schleinitz N, Boutiere-Albanese B, Camoin L, Bardin N, Guis S, Pouget J, Cognet C, Pellissier JF, Dignat-George F: Молекула адгезии тромбоцитов-эндотелиальных клеток-1 и CD146: растворимые уровни и in situ экспрессия молекул клеточной адгезии, участвующих в когезии эндотелиальных клеток при идиопатических воспалительных миопатиях.Дж. Ревматол 2006; 33:1623-1630.
- Volpi N, Pecorelli A, Lorenzoni P, Di Lazzaro F, Belmonte G, Agliano M, Cantarini L, Giannini F, Grasso G, Valacchi G: Антиангиогенная изоформа VEGF при воспалительных миопатиях. Медиаторы Inflamm 2013; 2013:219313.
- Loenneke JP, Thiebaud RS, Abe T, Manfro IG, Marin PJ: Острые упражнения с ограничением кровотока для лечения мышечной дистрофии Дюшенна: будут ли они эффективны? Фронт Физиол 2013;4:114.
- Джайн А., Шарма М.С., Саркар С., Бхатия Р., Сингх С., Ханда Р.: Повышенная экспрессия молекул клеточной адгезии при воспалительных миопатиях: диагностическая ценность и понимание патогенеза. Фолиа Нейропатол 2009;47:33-42.
- Okada Y, Jinnin M, Makino T, Kajihara I, Makino K, Honda N, Nakayama W, Inoue K, Fukushima S, Ihn H: MIRSNP rs24 miR-146a связан с поражением мышц при полимиозите/дерматомиозите.Int J Dermatol 2014; 53:300-304.
- Parkes JE, Day PJ, Chinoy H, Lamb JA: Роль микроРНК в идиопатических воспалительных миопатиях. Curr Opin Rheumatol 2015; 27: 608-615.
- Ленин Р., Санкарамурти А., Мохан В., Баласубраманьям М.: Измененный иммунометаболизм на границе повышенного стресса эндоплазматического ретикулума (ЭР) у пациентов с диабетом 2 типа.J Leukoc Biol 2015;98:615-622.
- Liu R, Yi B, Wei S, Yang WH, Hart KM, Chauhan P, Zhang W, Mao X, Liu X, Liu CG, Wang L: Ось FOXP3-miR-146-NF-kappaB и терапия предраковых поражений простаты . Рак Res 2015; 75: 1714-1724.
- Wang JH, Shih KS, Wu YW, Wang AW, Yang CR: Ингибиторы гистондеацетилазы увеличивают экспрессию микроРНК-146a и усиливают негативную регуляцию передачи сигналов интерлейкина-lbeta в синовиоцитах, подобных фибробластам при остеоартрите.Хрящевой остеоартроз 2013; 21:1987-1996.
- Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, Kalbfleisch JH, Gao X, Kao RL, Williams DL, Li C: Ослабление сердечной дисфункции при полимикробном сепсисе с помощью микроРНК-146a опосредуется через нацеливание на IRAKI и экспрессия TRAF6.Дж. Иммунол 2015; 195:672-682.
- Zou L, Feng Y, Chen YJ, Si R, Shen S, Zhou Q, Ichinose F, Scherrer-Crosbie M, Chao W: Toll-подобный рецептор 2 играет критическую роль в сердечной дисфункции во время полимикробного сепсиса. Crit Care Med 2010; 38: 1335-1342.
- Xia P, Fang X, Zhang ZH, Huang Q, Yan KX, Kang KF, Han L, Zheng ZZ: Нарушение регуляции miRNA146a по сравнению с IRAKI вызывает персистенцию IL-17 в псориатических поражениях кожи.Immunol Lett 2012;148:151-162.
- Wang Q, Bozack SN, Yan Y, Boulton ME, Grant MB, Busik JV: Регуляция воспаления сетчатки ритмической экспрессией MiR-146a в диабетической сетчатке. Invest Ophthalmol Vis Sci 2014; 55:3986-3994.
Автор Контакты
Youhong Jiang
Институт рака, Первая дочерняя больница Китайского медицинского университета,
No.155 Nanjing North Street, Heping District, Shenyang, Liaoning, 110001, (Китай)
Тел. +86 024-83232354, [email protected]
Информация о статье / публикации
Предварительный просмотр первой страницы
Принято: 28 сентября 2016 г.
Опубликовано онлайн: 25 ноября 2016 г.
Дата выпуска выпуска: декабрь 2016 г.
Количество печатных страниц: 13
Количество фигурок: 7
Количество столов: 3
ISSN: 1015-8987 (печать)
eISSN: 1421-9778 (онлайн)
Для получения дополнительной информации: https://www.karger.com/CPB
Лицензия открытого доступа / Дозировка препарата / Отказ от ответственности
Эта статья находится под лицензией Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND). Использование и распространение в коммерческих целях, а также любое распространение измененного материала требует письменного разрешения. Дозировка препарата: авторы и издатель приложили все усилия, чтобы гарантировать, что выбор препарата и дозировка, указанные в этом тексте, соответствуют текущим рекомендациям и практике на момент публикации.Тем не менее, в связи с продолжающимися исследованиями, изменениями в правительственных постановлениях и постоянным потоком информации, касающейся лекарственной терапии и реакций на лекарства, читателю настоятельно рекомендуется проверять вкладыш в упаковке для каждого лекарства на предмет любых изменений в показаниях и дозировке, а также для дополнительных предупреждений. и меры предосторожности. Это особенно важно, когда рекомендуемый агент является новым и/или редко используемым лекарственным средством. Отказ от ответственности: заявления, мнения и данные, содержащиеся в этой публикации, принадлежат исключительно отдельным авторам и участникам, а не издателям и редакторам.Появление рекламы и/или ссылок на продукты в публикации не является гарантией, одобрением или одобрением рекламируемых продуктов или услуг или их эффективности, качества или безопасности. Издатель и редактор(ы) отказываются от ответственности за любой ущерб, нанесенный людям или имуществу в результате любых идей, методов, инструкций или продуктов, упомянутых в содержании или рекламе.
Кишечный микробный метаболит триметиламин N-оксид усугубляет РТПХ, вызывая поляризацию макрофагов M1 у мышей | Кровь
Применение трансплантации аллогемопоэтических стволовых клеток (ТГСК; алло-ТГСК) ограничено из-за тяжелой реакции «трансплантат против хозяина» (РТПХ), которая является основной причиной безрецидивной смертности. 1,2 Схемы кондиционирования при алло-ТГСК вызывают повреждение ткани хозяина и активируют антигенпрезентирующие клетки хозяина, в основном макрофаги и дендритные клетки, для презентации антигенов хозяина донорским Т-клеткам и выработки цитокинов, стимулирующих Т-клетки. 3 Расширенные аллореактивные Т-клетки, в свою очередь, атакуют органы хозяина, включая кожу, печень и кишечник, а также систему кроветворения, что приводит к повреждению и дисфункции органов. 4
Сообщалось, что микробные метаболиты кишечника вызывают воспалительные реакции и коррелируют с воспалительными заболеваниями. 5-7 Изменения в составе микробиоты кишечника связаны с патологическими процессами, 8 , при которых микробиом кишечника генерирует широкий спектр биоактивных метаболитов, выступающих медиатором микробного воздействия на хозяина. 9,10 Таким образом, кишечный микробиом, подобно эндокринному органу, взаимодействует с дистальными органами через пути, зависящие от метаболизма, что оказывает всеобъемлющее влияние на различные физиологические или патологические события хозяина. 7,11
Недавние исследования показали, что микробные метаболические короткоцепочечные жирные кислоты (КЦЖК) защищают эпителиальные клетки кишечника от аллореактивных Т-клеточно-опосредованных повреждений после алло-ТГСК. 12 Введение бутирата способно увеличить выживаемость мышей с РТПХ. 12 Индол, продуцируемый триптофаном в кишечнике, дополнительно продемонстрировал защиту от РТПХ на мышиной модели посредством передачи сигналов интерферона I типа. 13
На самом деле, разнообразие человеческого микробиома и метаболома намного превышает сложность человеческого генома. 14 Пищевые ингредиенты, содержащие холин, фосфатидилхолин и карнитин, с высоким содержанием яиц, печени, молочных продуктов, арахиса и т. д., могут метаболизироваться в триметиламин (ТМА) и впоследствии превращаться флавинмонооксигеназами печени хозяина в ТМА N — оксид (ТМАО). 15 Сообщалось, что ТМАО, как еще один циркулирующий кишечный микробный метаболит, способен вызывать воспаление сосудов и эндотелиальную дисфункцию за счет образования и активации воспалительных процессов NLRP3 в эндотелиальных клетках. 16 Примечательно, что структурный аналог холина, 3,3-диметил-1-бутанол (ДМБ), нелетально ингибирует образование ТМА и снижает концентрацию ТМАО у мышей, получавших диету с высоким содержанием холина, 17 , что позволяет предположить, что DMB может служить потенциальным терапевтическим подходом к TMAO-стимулируемому воспалительному заболеванию. 18,19 Однако остается неизвестным, влияет ли ТМАО на патофизиологический процесс РТПХ.
В нашем исследовании мы выяснили, что ТМАО усиливает поляризацию макрофагов M1 посредством активации воспалительных процессов NLRP3. Поляризованные воспалительные макрофаги создают среду для мощного ответа Т-хелперов типа 1 (Th2) и Th27, который усугубляет реакцию РТПХ (РТПХ).
Клетки селезенкимышей C57BL/6 измельчали через нейлоновый фильтр для клеток с размером пор 40 мкм, промывали и окрашивали антителами против CD4 или CD8 (BioLegend).Очищенные Т-клетки CD4 + и CD8 + активировали связанными с планшетами анти-CD3 (0,25 мкг/мл; eBioscience) и анти-CD28 (0,25 мкг/мл; eBioscience) в среде для культивирования Т-клеток (DMEM; HyClone) в течение 24 часов и обрабатывали ТМАО (300 мкМ) или носителем в течение дополнительных 24 часов. Экспрессия информационной РНК (мРНК) STAT3, STAT4, STAT6, T-bet, интерлейкина-4 (IL-4) и RORγt в CD4 + Т-клетках и IL-2, IL-5, фактора некроза опухоли-α (TNF-α) и интерферон-γ (IFN-γ) в CD8 + Т-клетках.Включение 5-бром-2′-дезоксиуридина (BrdU) предназначалось для измерения пролиферации Т-клеток с использованием набора III для мечения и обнаружения BrdU (Sigma-Aldrich).
Окрашивание MitoSOX (Yeasen), MitoTracker Green (Beyotime) и MitoTracker Red CMXRos (Beyotime) в BMDM проводили в соответствии с инструкциями производителя. Вкратце, BMDM окрашивали индикатором митохондриального супероксида MitoSOX Red в течение 10 минут или MitoTracker в течение 30 минут.Для окрашивания антигеном клеточной поверхности клетки селезенки окрашивали изотиоцианатом флуоресцеина-CD11b, фикоэритрином-F4/80 и белком хлорофилла перидинина-Cy5.5-CD16/32 (BioLegend) при 4°C в течение 30 минут. Данные были получены в течение 2 часов после окрашивания на LSR Fortessa (BD Biosciences) и проведен анализ с использованием программного обеспечения FlowJo.
Большеберцовую кость, подвздошную кишку, толстую кишку, печень и кожу мышей собирали и фиксировали в течение ночи в фиксаторе периодат-лизин-параформальдегид.Фиксированные ткани или клетки окрашивали анти-CD3 (1:100; Abcam), tdTomato-распознаваемым анти-красным флуоресцентным белком (1:200; Abcam), анти-F4/80 (1:300; Abcam), анти- индуцируемая NO-синтаза (iNOS) (1:800; Abcam), анти-NLRP3 (1:100; Adipogen), антиапоптоз-ассоциированный пятнистый белок, содержащий CARD (ASC) (1:100; Affinity Biosciences), анти- –NF-κB (1:100; Proteintech) и анти-p-NF-κB (1:50; Affinity Biosciences). Окрашивание MitoSOX и JC-1 проводили в соответствии с инструкциями производителя (Beyotime, Китай).Подробный протокол рассматривается в дополнительных материалах и методах.
Чтобы определить, повлияет ли ТАМО на тяжесть РТПХ, мышам-реципиентам давали ТМАО за 2 недели до летального облучения для повышения уровня ТМАО в сыворотке (рис. 1А). По сравнению с реципиентами с обедненными Т-клетками (TCD)-BM (BMT) концентрация ТМАО в сыворотке была выше у реципиентов, которым переливали TCD-BM и селезеночные CD3 + T-клетки (TCD-BM+T, GVHD, 7.11 мкМ против 16,8 мкМ), а ежедневное пероральное лечение ТМАО значительно увеличивало концентрацию ТМАО в сыворотке либо у мышей с BMT, либо у мышей с РТПХ (рис. 1B). У реципиентов TCD-BM + T, получавших ТМАО, выживаемость и показатели РТПХ были значительно хуже, чем у мышей с РТПХ, получавших носитель (рис. 1C-D). На 14-й день гистопатологическое изображение показало более серьезное повреждение тканей-мишеней РТПХ (рис. 1E) и увеличение гистологических показателей в подвздошной кишке, толстой кишке, коже и печени (рис. 1F) у реципиентов РТПХ, получавших ТМАО.На репрезентативном изображении ткани реципиентов TCD-BM + T, получавших ТМАО, в тканях подвздошной кишки и толстой кишки наблюдались повышенная потеря крипт и изъязвление. Были обнаружены более тяжелая цитоплазматическая вакуолизация печени и повышенная плотность коллагена, а также атрофия серозного жира в коже (рис. 1Е; дополнительная рис. 1А). Эти данные показали, что ТМАО усугубляет тяжесть индуцированной Т-клетками GVHR при аллогенной ТГСК.
Рисунок 1.
ТМАО усугублял тяжесть и смертность мышей с РТПХ. (A) Экспериментальная схема протокола BMT или GVHD. Реципиенты BALB/c получали ТМАО или носитель с 14-го дня, а затем подвергались летальному облучению в 1-й день и инфузии донорских клеток C57BL/6 в 0-й день. Мышам вводили 5 × 10 6 TCD- Клетки КМ или 5 × 10 6 клеток TCD-BM и 2 × 10 6 селезеночных CD3 + Т-клеток соответственно. Данные были собраны на 14-й день, за исключением анализа кривой выживаемости на 50-й день.(B) Концентрация ТМАО в сыворотке определялась на 14-й день у мышей BMT (TCD-BM) и мышей с РТПХ (TCD-BM+T) с обработкой ТМАО или без нее. Данные были объединены из 3 независимых экспериментов. N = 8 в каждой группе. ** P < .01. (C) Кривая выживаемости Каплана-Мейера была определена у мышей BMT (n = 8) или у мышей с РТПХ (n = 28). График представляет объединенные данные из 2 (BMT) или 3 (GVHD) независимых экспериментов в присутствии или в отсутствие лечения TMAO. ** P < .01. (D) Оценки РТПХ были проанализированы у мышей BMT (n = 8) или мышей с РТПХ (n = 28).График представляет объединенные данные из 2 (BMT) или 3 (GVHD) независимых экспериментов в присутствии или в отсутствие лечения TMAO. ** P < .01. (E) Репрезентативное окрашивание тканей гематоксилином и эозином (H&E) подвздошной кишки, толстой кишки, кожи и печени мышей BMT или мышей GVHD. Масштабная линейка, 200 мкм. (F) Гистологический анализ признаков РТПХ в парафиновых срезах подвздошной кишки, толстой кишки, кожи и ткани печени проводили на мышах BMT или мышах с РТПХ, с лечением ТМАО или без него. N = 10 в каждой группе. Объединенные данные были из 3 независимых экспериментов.* P < 0,05; ** P < .01.
Рисунок 1.
ТМАО усугублял тяжесть и смертность мышей с РТПХ. (A) Экспериментальная схема протокола BMT или GVHD. Реципиенты BALB/c получали ТМАО или носитель с 14-го дня, а затем подвергались летальному облучению в 1-й день и инфузии донорских клеток C57BL/6 в 0-й день. Мышам вводили 5 × 10 6 TCD- Клетки КМ или 5 × 10 6 клеток TCD-BM и 2 × 10 6 селезеночных CD3 + Т-клеток соответственно.Данные были собраны на 14-й день, за исключением анализа кривой выживаемости на 50-й день. (B) Концентрация ТМАО в сыворотке определялась на 14-й день у мышей BMT (TCD-BM) и мышей с РТПХ (TCD-BM+T), с или без Лечение ТМАО. Данные были объединены из 3 независимых экспериментов. N = 8 в каждой группе. ** P < .01. (C) Кривая выживаемости Каплана-Мейера была определена у мышей BMT (n = 8) или у мышей с РТПХ (n = 28). График представляет объединенные данные из 2 (BMT) или 3 (GVHD) независимых экспериментов в присутствии или в отсутствие лечения TMAO.** P < .01. (D) Оценки РТПХ были проанализированы у мышей BMT (n = 8) или мышей с РТПХ (n = 28). График представляет объединенные данные из 2 (BMT) или 3 (GVHD) независимых экспериментов в присутствии или в отсутствие лечения TMAO. ** P < .01. (E) Репрезентативное окрашивание тканей гематоксилином и эозином (H&E) подвздошной кишки, толстой кишки, кожи и печени мышей BMT или мышей GVHD. Масштабная линейка, 200 мкм. (F) Гистологический анализ признаков РТПХ в парафиновых срезах подвздошной кишки, толстой кишки, кожи и ткани печени проводили на мышах BMT или мышах с РТПХ, с лечением ТМАО или без него.N = 10 в каждой группе. Объединенные данные были из 3 независимых экспериментов. * P < 0,05; ** P < .01.
Чтобы оценить влияние диеты с высоким содержанием холина на тяжесть РТПХ, мышей кормили контрольной диетой или диетой с добавлением холина с -14 дня, а затем в присутствии или отсутствии ДМБ в питьевой воде (рис. 2А). . Холиновая диета заметно повышала концентрацию ТМАО в сыворотке у мышей с РТПХ с 16 лет.от 59 мкМ до 62,14 мкМ, однако добавление ДМБ в питьевую воду значительно снизило концентрацию ТМАО в сыворотке до 22,27 мкМ (рис. 2В). Реципиенты TCD-BM + T на диете с добавлением холина показали значительно укороченную кривую выживаемости (рис. 2C) и повышенный рейтинг РТПХ (рис. 2D; * P < 0,05, ** P < 0,01, TCD -BM+T против TCD-BM+T+холин). Примечательно, что пероральный прием ДМБ снижал тяжесть холиноиндуцированной РТПХ (рис. 2C-D; # P <0,05, ## P <0,05).01, ТЦД-БМ+Т+холин по сравнению с ТЦД-БМ+Т+холин+ДМБ). Однако лечение DMB не оказало существенного влияния на тяжесть РТПХ и кривую выживаемости в отсутствие добавок холина (рис. 2C-D). Кроме того, гистопатологический анализ на 14-й день показал более тяжелую РТПХ в органах мышей, получавших холин (рис. 2Е), с повышенными гистологическими показателями в подвздошной кишке, толстой кишке, коже и печени (рис. 2F), в то время как замена ДМБ в питьевой воде предотвращала выброс холина. - усиленное гистопатологическое повреждение (рис. 2E-F). На репрезентативном изображении мышей с РТПХ, получавших ДМБ и получавших холин, ткани подвздошной и толстой кишки показали уменьшение изъязвления, где воспалительные клетки в печени были менее инфильтрированы, а фолликулы и адипоциты в коже были восстановлены (рис. 2Е; дополнительный рисунок 2А). ).Наши результаты показали, что повышенная тяжесть РТПХ, наблюдаемая на холиновой диете, связана с образованием ТМАО.
Рисунок 2.
ДМБ снижал ТМАО, продуцируемый диетой с высоким содержанием холина, и уменьшал повреждение тканей у мышей с РТПХ, получавших холин. (A) Экспериментальная схема мышей BMT и протокол мышей с РТПХ. Реципиенты BALB/c получали DMB, холинсодержащую диету или холин+DMB с -14 дня, после чего подвергались летальному облучению в день -1 и переливались донорскими клетками C57BL/6 в день 0.Данные были собраны на 14-й день, за исключением анализа кривой выживания на 50-й день. (B) Концентрация ТМАО в сыворотке определялась у мышей с РТПХ, получавших диету с DMB, холином или холином и DMB. Данные были объединены из 3 независимых экспериментов. N = 7 в каждой группе. ** P < .01. (C) Кривая выживаемости Каплана-Мейера была определена у мышей с РТПХ, получавших носитель (n = 25), DMB (n = 20), холин (n = 25) или диету холин + DMB (n = 25). На графике представлены объединенные данные из 2 (DMB) или 3 (носитель, холин, холин+DMB) независимых экспериментов.** P < ,01 (TCD-BM+T против TCD-BM+T+холин). ## P < ,01 (TCD-BM+T+холин против TCD-BM+T+холин+DMB). (D) Оценка РТПХ была определена у мышей с РТПХ, получавших носитель (n = 25), ДМБ (n = 20), холин (n = 25) или холин + ДМБ (n = 25). На графике представлены объединенные данные из 2 (DMB) или 3 (носитель, холин, холин+DMB) независимых экспериментов. * P < ,05, ** P <,01 (TCD-BM+T против TCD-BM+T+холин). # P < .05, ## P < ,01 (TCD-BM+T+холин против TCD-BM+T+холин+DMB). (E) Репрезентативное окрашивание H&E тканей подвздошной кишки, толстой кишки, кожи и печени у мышей с РТПХ, которых кормили холином, DMB или холином и диетой DMB. Масштабная линейка, 200 мкм. (F) Гистологический анализ парафиновых срезов подвздошной кишки, толстой кишки, кожи и печени мышей с РТПХ проводили на 14-й день. * P <,05, ** P <,01. Данные были объединены из 3 независимых экспериментов. N = 10 в каждой группе. (G) Было проанализировано бактериальное разнообразие фекальной микробиоты мышей с РТПХ, получавших диету с высоким содержанием холина или контрольную диету.N = 6 в каждой группе. (H) Анализ основных координат расстояний сообществ фекальной микробиоты между диетой с высоким содержанием холина и диетой с едой у мышей с РТПХ. N = 6 в каждой группе. (I) Линейный дискриминантный анализ в сочетании с измерениями размера эффекта для выявления микробных таксонов кишечника у мышей с РТПХ, получавших диету с высоким содержанием холина или контрольную диету. N = 6 в каждой группе. нс, не значимо; ПК, основной компонент.
Рисунок 2.
ДМБ снижал уровень ТМАО, продуцируемого в рационе с высоким содержанием холина, и уменьшал повреждение тканей у мышей с РТПХ, получавших холин. (A) Экспериментальная схема мышей BMT и протокол мышей с РТПХ. Реципиентам BALB/c давали DMB, холинсодержащую диету или холин+DMB с -14 дня, а затем подвергали летальному облучению в день -1 и переливали донорские клетки C57BL/6 в день 0. Данные собирали на 14-й день, за исключением анализа кривой выживания на 50-й день. (B) Концентрацию ТМАО в сыворотке определяли у мышей с РТПХ, получавших диету с DMB, холином или холином и DMB. Данные были объединены из 3 независимых экспериментов. N = 7 в каждой группе.** P < .01. (C) Кривая выживаемости Каплана-Мейера была определена у мышей с РТПХ, получавших носитель (n = 25), DMB (n = 20), холин (n = 25) или диету холин + DMB (n = 25). На графике представлены объединенные данные из 2 (DMB) или 3 (носитель, холин, холин+DMB) независимых экспериментов. ** P < ,01 (TCD-BM+T против TCD-BM+T+холин). ## P < ,01 (TCD-BM+T+холин против TCD-BM+T+холин+DMB). (D) Оценка РТПХ была определена у мышей с РТПХ, получавших носитель (n = 25), ДМБ (n = 20), холин (n = 25) или холин + ДМБ (n = 25).На графике представлены объединенные данные из 2 (DMB) или 3 (носитель, холин, холин+DMB) независимых экспериментов. * P < ,05, ** P <,01 (TCD-BM+T против TCD-BM+T+холин). # P < ,05, ## P < ,01 (ТЦД-БМ+Т+холин против ТЦД-БМ+Т+холин+ДМБ). (E) Репрезентативное окрашивание H&E тканей подвздошной кишки, толстой кишки, кожи и печени у мышей с РТПХ, которых кормили холином, DMB или холином и диетой DMB. Масштабная линейка, 200 мкм. (F) Гистологический анализ парафиновых срезов подвздошной кишки, толстой кишки, кожи и тканей печени мышей с РТПХ проводили на 14-й день.* Р < ,05, ** Р < ,01. Данные были объединены из 3 независимых экспериментов. N = 10 в каждой группе. (G) Было проанализировано бактериальное разнообразие фекальной микробиоты мышей с РТПХ, получавших диету с высоким содержанием холина или контрольную диету. N = 6 в каждой группе. (H) Анализ основных координат расстояний сообществ фекальной микробиоты между диетой с высоким содержанием холина и диетой с едой у мышей с РТПХ. N = 6 в каждой группе. (I) Линейный дискриминантный анализ в сочетании с измерениями размера эффекта для выявления микробных таксонов кишечника у мышей с РТПХ, получавших диету с высоким содержанием холина или контрольную диету.N = 6 в каждой группе. нс, не значимо; ПК, основной компонент.
Таким образом, мы проанализировали, может ли диета с высоким содержанием холина у мышей с РТПХ изменить микробиоту кишечника. Интересно, что между мышами, получавшими высокое содержание холина, и мышами, получавшими контрольную диету, не наблюдалось существенных различий в бактериальном разнообразии и богатстве (рис. 2G). Основной координатный анализ расстояний (рис. 2H) и фекальных бактериальных сообществ (дополнительный рисунок 2B) был одинаковым в обеих группах.Среди них 9 таксонов микробов кишечника были идентифицированы в 2 группах с помощью линейного дискриминантного анализа в сочетании с измерениями размера эффекта, где Anaeroplasmatales, Anaeroplasmataceae и Anaeroplasma были снижены у мышей, получавших холин, а также Deferribacterales, Deferribacteres, Mucispirillum, Deferribacteraceae, Deferribacteres и AF12. были увеличены у мышей, получавших контрольную диету (рис. 2I).
Затем мы попытались изучить потенциальные механизмы, способствующие обострению РТПХ, вызванному ТМАО.Во-первых, поскольку костный мозг и селезенка являются гематолимфоидными органами и мишенями РТПХ, было проанализировано 23-25 аллогенных инфильтратов внутри костного мозга и внутри селезенки Т-клеток. С помощью прижизненной флуоресцентной визуализации костных мозгов черепа было обнаружено, что усиленные клетки GFP + (eGFP + ) TCD-BM приживаются в костный мозг на 7-й день либо у мышей, получавших носитель, либо у мышей, получавших ТМАО, и росли как массивная колония на 14-й день (рис. 3A-B). Однако у мышей с РТПХ, которым вводили TCD-BM и Т-клетки, было обнаружено, что Т-клетки tdTomato + (красные) конкурируют с BM за счет снижения приживления клеток BM (рис. 3B, нижняя панель).Пероральное лечение ТМАО значительно усиливало прижизненный рост Т-клеток tdTomato + и снижало пролиферацию клеток eGFP + BM у мышей с РТПХ в течение 2 недель наблюдения (дополнительная фигура 3A-B), где разница флуоресценции была не столь значительной. у мышей, которым вводили только клетки TCD-BM (рис. 3B). Эта усиленная ТМАО инфильтрация Т-клеток в BM также была подтверждена иммунофлуоресцентной визуализацией ex vivo (рис. 3C; дополнительная рис. 3C) и анализом образцов бедренной кости с помощью флуоресцентно-активированного клеточного сортера (FACS) (дополнительная рис. 3D).
Рисунок 3.
Дифференцировка Th2 и Th27 наблюдалась у мышей с РТПХ, получавших ТМАО, но не в системе культивирования in vitro, получавшей ТМАО. (A) Экспериментальная схема мышей BMT или протокол мышей GVHD. Реципиентов кормили ТМАО или носителем с -14 дня и далее. Данные были собраны на 7-й и 14-й день. (B) Репрезентативное флуоресцентное изображение in vivo черепа BM у мышей-реципиентов на 7-й и 14-й день.Реципиентам BALB/c вводили клетки B6-Tg (CAG-EGFP) TCD-BM для создания модели BMT или клетки B6-Tg (CAG-EGFP) TCD-BM и B6.129(Cg)- Gt(ROSA)26 Sortm4(ACTB-tdTomato-EGFP)/Nju (mT/mG) селезеночные CD3 + T-клетки для получения мышей GVHD. Были обнаружены привитые клетки tdTomato + T (красные) и клетки GFP + TCD-BM (зеленые). Прижизненные сосуды окрашивали декстраном-Cy5 (синий) путем инъекции в хвостовую вену. Масштабная линейка, 200 мкм. (C-E) Клетки TCD-BM от мышей C57BL/6 с Т-клетками селезенки от B6.129(Cg)-Gt(ROSA)26 Sortm4(ACTB-tdTomato-EGFP)/Nju (mT/mG) мышам внутривенно вводили совместно летально облученным мышам BALB/c. (C) Репрезентативное изображение аллогенных tdTomato + Т-клеток в бедренной кости мышей, окрашенных распознаваемым tdTomato антителом против красного флуоресцентного белка (красный, Alexa Fluor 594) и антителом против CD3 (зеленый, Alexa Fluor 488). Клеточные ядра окрашивали 4′,6-диамидино-2-фенилиндолом (DAPI). Масштабная линейка, 100 мкм. (D) Репрезентативное прижизненное изображение селезенки реципиентов in vivo на 7-й и 14-й день.Привитые Т-клетки tdTomato + (красные) и сосудистую сеть (синие) визуализировали в присутствии или в отсутствие обработки ТМАО. Масштабная линейка, 200 мкм. (E) Экспрессия мРНК IFN-γ (n = 6), IL-4 (n = 6), IL-17 (колонка слева направо, n = 5, 4), T-bet (n = 6), Определяли RORγt (столбец слева направо, n = 5, 6) и STAT3 (n = 6), STAT4 (n = 4), STAT6 (n = 7) в аллогенных селезеночных tdTomato + T-клетках. Т-клетки выделяли из селезенки при РТПХ на 14-й день с обработкой ТМАО или без нее. Данные были объединены из 2 независимых экспериментов.* Р < ,05, ** Р < ,01. (F) Т-клетки CD4 + , выделенные от мышей C57BL/6, стимулировали антителами против CD3 и против CD28 в присутствии или в отсутствие обработки ТМАО (300 мкМ). Экспрессию мРНК IL-4, IL-17, IFN-γ, Gata3 и STAT3, STAT4, STAT6 определяли через 24 часа после обработки ТМАО. N = 3 в каждой группе.
Рисунок 3.
Дифференцировка Th2 и Th27 наблюдалась у мышей с РТПХ, получавших ТМАО, но не в системе культивирования in vitro, получавшей ТМАО. (A) Экспериментальная схема мышей BMT или протокол мышей GVHD. Реципиентов кормили ТМАО или носителем с -14 дня и далее. Данные были собраны на 7-й и 14-й день. (B) Репрезентативное флуоресцентное изображение костного мозга черепа in vivo у мышей-реципиентов на 7-й и 14-й день. Реципиентам BALB/c вводили B6-Tg (CAG-EGFP) TCD. -BM клетки для создания модели BMT или B6-Tg (CAG-EGFP) клетки TCD-BM и B6.129(Cg)-Gt(ROSA)26 Sortm4(ACTB-tdTomato-EGFP)/Nju (mT /mG) CD3 + Т-клеток селезенки для получения мышей с РТПХ.Были обнаружены привитые клетки tdTomato + T (красные) и клетки GFP + TCD-BM (зеленые). Прижизненные сосуды окрашивали декстраном-Cy5 (синий) путем инъекции в хвостовую вену. Масштабная линейка, 200 мкм. (CE) Клетки TCD-BM от мышей C57BL/6 с Т-клетками селезенки от мышей B6.129(Cg)-Gt(ROSA)26 Sortm4(ACTB-tdTomato-EGFP)/Nju (mT/mG) вводили совместно внутривенно у летально облученных мышей BALB/c. (C) Репрезентативное изображение аллогенных tdTomato + Т-клеток в бедренной кости мышей, окрашенных распознаваемым tdTomato антителом против красного флуоресцентного белка (красный, Alexa Fluor 594) и антителом против CD3 (зеленый, Alexa Fluor 488).Клеточные ядра окрашивали 4′,6-диамидино-2-фенилиндолом (DAPI). Масштабная линейка, 100 мкм. (D) Репрезентативное прижизненное изображение селезенки реципиента in vivo на 7-й и 14-й день. Привитые tdTomato + Т-клетки (красный) и сосудистая сеть (синий) были визуализированы в присутствии или в отсутствие лечения ТМАО. Масштабная линейка, 200 мкм. (E) Экспрессия мРНК IFN-γ (n = 6), IL-4 (n = 6), IL-17 (колонка слева направо, n = 5, 4), T-bet (n = 6), Определяли RORγt (столбец слева направо, n = 5, 6) и STAT3 (n = 6), STAT4 (n = 4), STAT6 (n = 7) в аллогенных селезеночных tdTomato + T-клетках.Т-клетки выделяли из селезенки при РТПХ на 14-й день с обработкой ТМАО или без нее. Данные были объединены из 2 независимых экспериментов. * Р < ,05, ** Р < ,01. (F) Т-клетки CD4 + , выделенные от мышей C57BL/6, стимулировали антителами против CD3 и против CD28 в присутствии или в отсутствие обработки ТМАО (300 мкМ). Экспрессию мРНК IL-4, IL-17, IFN-γ, Gata3 и STAT3, STAT4, STAT6 определяли через 24 часа после обработки ТМАО. N = 3 в каждой группе.
Мы дополнительно адаптировали наш протокол визуализации для отслеживания динамики Т-клеток в РТПХ селезенки in vivo.Частота Т-клеток tdTomato + постепенно увеличивалась в селезенке аллогенных реципиентов с 7-го по 14-й день (рис. 3D; дополнительная рис. 3E), где введение ТМАО заметно повышало рост Т-клеток tdTomato + в селезенке. синусоиды в процентах 60,6% ± 3,5% против 44,1% ± 1,8% (рис. 3D; дополнительный рисунок 3E-F). Кроме того, анализ FACS показал повышенную инфильтрацию Т-клеток tdTomato + в подвздошной кишке (дополнительный рисунок 3G), толстой кишке (дополнительный рисунок 3H) и печени (дополнительный рисунок 3I) у реципиентов РТПХ, получавших ТМАО.
Для выяснения механизма, с помощью которого ТМАО усиливает аллогенную стимуляцию Т-клеток при РТПХ, определяли экспрессию мРНК в различных популяциях Th-клеток, включая Th2-связанные T-bet и STAT4, Th3-связанные IL-4 и STAT6, а также как Th27-связанные STAT3 и RORγt. 26 Привитые tdTomato + Т-клетки выделяли из селезенки РТПХ с обработкой ТМАО или без нее, а затем помещали в систему выделения РНК.Анализом ПЦР в реальном времени было обнаружено, что экспрессия IFN-γ, IL-17, T-bet, STAT4, STAT3 и RORγt в Т-клетках tdTomato + повышалась у получавших ТМАО реципиентов, тогда как IL-4 и STAT6 повышались. без изменений (рис. 3E). Затем мы обработали изолированные Т-клетки CD4 + и CD8 + ТМАО в чашке для культивирования, чтобы дополнительно исследовать, стимулирует ли ТМАО функцию Th2 и Th27 in vitro. Неожиданно ни повышенная пролиферация Т-клеток, измеренная по включению BrdU (дополнительная фигура 3J-K), ни экспрессия факторов транскрипции и цитокинов, связанных с РТПХ (рисунок 3F; дополнительная фигура 3L), не наблюдались в обработанных ТМАО CD4 + и CD8. + Т-клетки.Таким образом, влияние ТМАО на индукцию аллогенной экспансии Т-клеток, а также на ответ Th2 и Th27 наблюдали не в системе культивирования in vitro, а у мышей с РТПХ in vivo.
Учитывая, что in vivo ответ Th2 и Th27 требует поляризации макрофагов M1, служащей антигенпрезентирующими признаками, 27,28 мы дополнительно отсортировали F4/80 + CD11b + мышиные макрофаги (дополнительный рисунок 4A) и CD16/32 + популяция M1 из F4/80 + CD11b + макрофагов (рис. 4А) из селезенки при РТПХ для исследования дифференцировки макрофагов.Примечательно, что по сравнению с мышами с РТПХ, получавшими носитель, количество макрофагов в селезенке F4/80 + CD11b + было значительно увеличено у реципиентов TCD-BM+T, подвергнутых лечению ТМАО, где процент F4/80 + CD11b + CD16/32 + Макрофаги M1 по сравнению со всей популяцией F4/80 + CD11b + были выше у мышей с РТПХ, получавших ТМАО (рис. 4A-B). Иммунофлуоресценция органов-мишеней РТПХ, включая подвздошную кишку, толстую кишку и печень, показала более сильную инфильтрацию макрофагов F4/80 + с фенотипом M1 (F4/80 + iNOS + ) у мышей с РТПХ, получавших ТМАО (рис. 4C; дополнительный рисунок 4B-D).
Рисунок 4.
Обработка ТМАО усиливала инфильтрацию макрофагов и вызывала поляризацию M1 как in vitro, так и in vivo. (A-E) Летально облученным реципиентам BALB/c вводили TCD-BM и селезеночные CD3 + Т-клетки от доноров C57BL/6. (A) Селезеночная популяция CD16/32 + M1 была отсортирована от макрофагов F4/80 + CD11b + у мышей с БТПХ на 14-й день в присутствии или в отсутствие лечения ТМАО.(B) Определен процент селезеночного CD16/32 + M1 фенотипа из F4/80 + CD11b + макрофагов. Данные были объединены из 2 независимых экспериментов. N = 9 в каждой группе. ** P < .01. ● ТЦД-БМ+Т; ▪, ТЦД-БМ+Т+ТМАО. (C) Репрезентативное изображение инфильтративных макрофагов M1 F4/80 + (красный, Alexa Fluor 594) iNOS + (зеленый, Alexa Fluor 488) в тканях подвздошной кишки, толстой кишки и печени от реципиентов РТПХ с лечением ТМАО или без него.Клеточные ядра окрашивали DAPI. Масштабная линейка, 10 мкм. (D-E) Клетки селезенки F4/80 + были отсортированы от мышей с РТПХ. (D) Экспрессию IL-6, CXCL9 и CXCL10 определяли с помощью ПЦР с обратной транскрипцией (ОТ-ПЦР). Данные были объединены из 3 независимых экспериментов. N = 6 в каждой группе. ** P < .01. (E) Экспрессия NLRP3, IL-1β и расщепленного IL-1β определялась вестерн-блоттингом. (F) Анализировали экспрессию IL-1β (n = 6), IL-6 (n = 3), TNF-α (n = 6), CXCL9 (n = 5) и CXCL10 (n = 5) в BMDM. с помощью ОТ-ПЦР в присутствии или в отсутствие обработки ТМАО (300 мкМ).** P < .01. (G) Цитокины, высвобождаемые в супернатанты BMDM, включая IL-1β (n = 4), IL-6 (столбец слева направо, n = 5, 4) и TNF-α (столбец слева направо, n = 3, 4) , были обнаружены с помощью твердофазного иммуноферментного анализа (ELISA) после обработки ТМАО (300 мкМ). ** P < .01.
Рисунок 4.
Обработка ТМАО усиливала инфильтрацию макрофагов и вызывала поляризацию M1 как in vitro, так и in vivo. (A-E) Летально облученным реципиентам BALB/c вводили TCD-BM и селезеночные CD3 + Т-клетки от доноров C57BL/6.(A) Селезеночная популяция CD16/32 + M1 была отсортирована от макрофагов F4/80 + CD11b + у мышей с БТПХ на 14-й день в присутствии или в отсутствие лечения ТМАО. (B) Определен процент селезеночного CD16/32 + M1 фенотипа из F4/80 + CD11b + макрофагов. Данные были объединены из 2 независимых экспериментов. N = 9 в каждой группе. ** P < .01. ● ТЦД-БМ+Т; ▪, ТЦД-БМ+Т+ТМАО. (C) Репрезентативное изображение инфильтративных макрофагов M1 F4/80 + (красный, Alexa Fluor 594) iNOS + (зеленый, Alexa Fluor 488) в тканях подвздошной кишки, толстой кишки и печени от реципиентов РТПХ с лечением ТМАО или без него.Клеточные ядра окрашивали DAPI. Масштабная линейка, 10 мкм. (D-E) Клетки селезенки F4/80 + были отсортированы от мышей с РТПХ. (D) Экспрессию IL-6, CXCL9 и CXCL10 определяли с помощью ПЦР с обратной транскрипцией (ОТ-ПЦР). Данные были объединены из 3 независимых экспериментов. N = 6 в каждой группе. ** P < .01. (E) Экспрессия NLRP3, IL-1β и расщепленного IL-1β определялась вестерн-блоттингом. (F) Анализировали экспрессию IL-1β (n = 6), IL-6 (n = 3), TNF-α (n = 6), CXCL9 (n = 5) и CXCL10 (n = 5) в BMDM. с помощью ОТ-ПЦР в присутствии или в отсутствие обработки ТМАО (300 мкМ).** P < .01. (G) Цитокины, высвобождаемые в супернатанты BMDM, включая IL-1β (n = 4), IL-6 (столбец слева направо, n = 5, 4) и TNF-α (столбец слева направо, n = 3, 4) , были обнаружены с помощью твердофазного иммуноферментного анализа (ELISA) после обработки ТМАО (300 мкМ). ** P < .01.
Фенотип макрофагов M1 характеризуется активацией NLRP3 29 и индукцией провоспалительных медиаторов, включая IL-1β, IL-6, TNF-α, CXCL9 и CXCL10. 30 В нашем исследовании экспрессия характерных цитокинов M1 IL-6, CXCL9, CXCL10 и IL-1β или гена Nlrp3 была повышена в макрофагах F4/80 + селезенки, полученных от мышей с БТПХ, получавших ТМАО (рис. 4D-Е). Все эти данные указывают на возможность того, что ТМАО может косвенно вызывать дифференцировку Th2 и Th27 посредством регуляции поляризации макрофагов.
Для дальнейшего изучения функции TMAO на поляризацию макрофагов мы оценили характеристики BMDM in vitro в присутствии или в отсутствие TMAO.После стимуляции ТМАО либо экспрессия мРНК, либо продукция белков M1-характеризуемых IL-1β, IL-6, TNF-α, CXCL9 и CXCL10 значительно увеличивались при BMDM (рис. 4F-G), что позволяет предположить в нашем исследовании, что TMAO индуцирует поляризацию макрофагов до фенотипа М1.
Далее мы исследовали, участвует ли инфламмасома NLRP3, ключевой белковый комплекс в поляризации M1, в ответе BMDM на TMAO.Мы обнаружили, что ASC, который был дисперсно распределен в контрольных покоящихся макрофагах, поляризован на 1 сторону перинуклеарной области в ответ на стимуляцию ТМАО (рис. 5А; дополнительный рисунок 5А). Экспрессия NLRP3 была увеличена и частично локализована с ASC в околоядерной области в BMDM, обработанных TMAO (рис. 5A-B). Соответственно, продукция расщепленного IL-1β индуцировалась в BMDM, обработанных ТМАО, где холин не влиял на экспрессию NLRP3 и продукцию IL-1β in vitro (рис. 5B). CY-09, ингибитор активации NLRP3, связываясь с сайтом связывания аденозинтрифосфата NACHT-домена NLRP3, заметно подавлял выработку и поляризованное накопление расщепленной каспазы-1, связанной с ТМАО (рис. 5C-E; дополнительная рис. 5B), как а также секрецию IL-1β (рис. 5F).Кроме того, индуцированная ТМАО экспрессия мРНК характерных цитокинов или хемокинов M1, включая IL-1β, IL-6, TNF-α, CXCL9 и CXCL10, подавлялась CY-09 (рис. 5G). Соответственно, Nlrp3 -/- BMDM не демонстрировали усиленного профиля характерной экспрессии M1 (рис. 5H), что указывает на то, что активация NLRP3 играет ключевую роль в поляризации макрофагов, обработанных ТМАО.
Рисунок 5.
ТМАО усиливает поляризацию M1 посредством активации инфламмасомы NLRP3. (A) Репрезентативное иммунофлуоресцентное окрашивание NLRP3 (зеленый, Alexa Fluor 488), ASC (красный, Alexa Fluor 594), DAPI (синий) на BMDM после стимуляции TMAO (300 мкМ) в течение 24 часов. Масштабная линейка, 5 мкм. (B) Вестерн-блоттинг анализ NLRP3, IL-1β и расщепленного IL-1β BMDM, культивируемых с TMAO (300 мкМ) или холином (300 мкМ) в течение 24 часов. (CG) BMDM культивировали с ТМАО (300 мкМ) в присутствии или в отсутствие CY-09 (1 мкМ) в течение 24 часов. (C) Активность Casapse-1 (N = 5 в каждой группе; ** P <.01). (D) Репрезентативное изображение FLICA Casapse-1, которое связывается только с активированной каспазой-1 (зеленый, FAM FLICA, синий, DAPI). Масштабная линейка, 5 мкм. (E) Был определен анализ проточной цитометрии FLICA Casapse-1. (F) Концентрации IL-1β в супернатанте BMDM, культивируемых с TAMO или TMAO+CY-09, определяли с помощью ELISA. N = 4 в каждой группе. ** P < .01. (G) Экспрессия IL-1β (столбец слева направо, n = 4, 3, 4), IL-6 (столбец слева направо, n = 3, 4, 3), TNF-α (столбец слева направо, n = 3, 3, 5), CXCL9 (столбец слева направо, n = 4, 3, 3) и CXCL10 (столбец слева направо, n = 3, 3, 6) в BMDM, стимулированных ТМАО или ТМАО + CY -09 определяли с помощью ОТ-ПЦР.* Р < ,05, ** Р < ,01. (H) Определена экспрессия IL-1β, IL-6, TNF-α, CXCL9 и CXCL10 в Nlrp3 -/- BMDM или WT BMDM. N = 3 в каждой группе.
Рисунок 5.
ТМАО усиливает поляризацию M1 посредством активации инфламмасомы NLRP3. (A) Репрезентативное иммунофлуоресцентное окрашивание NLRP3 (зеленый, Alexa Fluor 488), ASC (красный, Alexa Fluor 594), DAPI (синий) на BMDM после стимуляции TMAO (300 мкМ) в течение 24 часов.Масштабная линейка, 5 мкм. (B) Вестерн-блоттинг анализ NLRP3, IL-1β и расщепленного IL-1β BMDM, культивируемых с TMAO (300 мкМ) или холином (300 мкМ) в течение 24 часов. (CG) BMDM культивировали с ТМАО (300 мкМ) в присутствии или в отсутствие CY-09 (1 мкМ) в течение 24 часов. (C) Активность Casapse-1 (N = 5 в каждой группе; ** P <,01). (D) Репрезентативное изображение FLICA Casapse-1, которое связывается только с активированной каспазой-1 (зеленый, FAM FLICA, синий, DAPI). Масштабная линейка, 5 мкм. (E) Был определен анализ проточной цитометрии FLICA Casapse-1.(F) Концентрации IL-1β в супернатанте BMDM, культивируемых с TAMO или TMAO+CY-09, определяли с помощью ELISA. N = 4 в каждой группе. ** P < .01. (G) Экспрессия IL-1β (столбец слева направо, n = 4, 3, 4), IL-6 (столбец слева направо, n = 3, 4, 3), TNF-α (столбец слева направо, n = 3, 3, 5), CXCL9 (столбец слева направо, n = 4, 3, 3) и CXCL10 (столбец слева направо, n = 3, 3, 6) в BMDM, стимулированных ТМАО или ТМАО + CY -09 определяли с помощью ОТ-ПЦР. * P < .05, ** P < .01. (H) Определена экспрессия IL-1β, IL-6, TNF-α, CXCL9 и CXCL10 в Nlrp3 -/- BMDM или WT BMDM. N = 3 в каждой группе.
Затем мы исследовали функцию ТМАО в отношении митохондриальной дисфункции, которая впоследствии способствует активации воспаления NLRP3. 31 Стимуляция ТМАО вызывала интенсивную продукцию митохондриальных активных форм кислорода (АФК) в BMDM.И иммунофлуоресценция, и анализ FACS показали, что митохондриальные АФК, меченные MitoSOX, увеличивались после обработки ТМАО (рис. 6А-С). Чтобы проверить, связано ли увеличение митохондриальных АФК с накоплением дисфункциональных митохондрий, было использовано окрашивание JC-1 для определения потенциала митохондриальной мембраны, а также были использованы 2 типа митохондриально-специфических флуоресцентных меток, чтобы отличить дышащие митохондрии (MitoTracker Red) от всех митохондрий. (МитоТрекер Грин). Стимуляция ТМАО вызвала снижение интенсивности флуоресценции красного JC-1 и увеличение интенсивности флуоресценции зеленого JC-1, что указывает на то, что потенциал митохондриальной мембраны рухнул после обработки ТМАО (рис. 6D; дополнительная рис. 6А).Кроме того, в BMDM, стимулированных ТМАО, наблюдалось накопление дисфункциональных митохондрий (MitoTracker Green high MitoTracker Red low ) (рис. 6E). Напротив, обработка макрофагов Mito-Tempo, продуцируемым митохондриями поглотителем АФК, снижала индуцированную ТМАО продукцию митохондриальных АФК (дополнительный рисунок 6B-C) и обращала вспять накопленную ASC активацию NLRP3-поляризованного (рисунок 6F; дополнительный рисунок 6D). Повышенная продукция IL-1β (рис. 6G-H) и каспазы-1 (рис. 6I-J) в макрофагах, обработанных ТМАО, также была отменена Mito-Tempo.Кроме того, Mito-Tempo блокировала связанную с ТМАО индукцию сигнатурных цитокинов макрофагов M1 (TNF-α и IL-6) или хемокинов (CXCL9 и CXCL10) (рис. 6K-L).
Рисунок 6.
ТМАО активировал NLRP3 через митохондриальные АФК. (A) Репрезентативное изображение BMDM, помеченных MitoSOX для обнаружения митохондриальных АФК (зеленый, Mito; красный, MitoSOX; синий, Hoechst). Масштабная линейка, 10 мкм. (B-E) Средняя интенсивность флуоресценции (MFI) MitoSOX, определенная с помощью ImageJ (B; N = 5 в каждой группе; ** P <.01) и процент экспрессирующих флуоресценцию MitoSOX, определенный с помощью анализа FACS (C) в BMDM, обработанных ТМАО. Потенциал митохондриальной мембраны как отношение красного JC-1 к зеленому JC-1 определяли с помощью иммунофлуоресценции (D; масштабная шкала, 10 мкм), а дисфункциональное митохондриальное дыхание, помеченное MitoTracker Green , высокое MitoTracker Red , низкое , определяли с помощью FACS. анализ (E) в BMDM, обработанных 300 мкМ TMAO в течение 24 часов. (F) Репрезентативное иммунофлуоресцентное окрашивание NLRP3 (зеленый, Alexa Fluor 488), ASC (красный, Alexa Fluor 594), DAPI (синий) в BMDM, обработанных TMAO или TMAO + Tempo.Масштабная линейка, 5 мкм. (G) Вестерн-блоттинг анализ NLRP3, IL-1β и расщепленного IL-1β в BMDM, обработанных TMAO или TMAO + Tempo. (H) Секретируемый IL-1β в супернатантах, культивируемых BMDM, с TMAO, Tempo или TMAO+Tempo. N = 4 в каждой группе. ** P < .01. (IJ) Активность каспазы-1, определяемая как концентрация pNA (I; N = 4 в каждой группе; * P < ,05, ** P <,01) или процент изотиоцианата флуоресцеина-FLICA (J) анализировали в BMDM, обработанные ТМАО или обработанные ТМАО + Tempo.(K) Экспрессию TNF-α (N = 3 в каждой группе) и IL-6 (N = 4 в каждой группе) определяли с помощью ELISA в супернатантах BMDM, культивируемых с TMAO, Tempo, TMAO+Tempo в течение 24 часов. ** P < .01. (L) Экспрессию CXCL9 и CXCL10 в BMDM определяли с помощью RT-PCR после обработки Tempo, TMAO или TMAO+Tempo в течение 24 часов. N = 3 в каждой группе. ** P < .01.
Рисунок 6.
ТМАО активировал NLRP3 через митохондриальные АФК. (A) Репрезентативное изображение BMDM, помеченных MitoSOX для обнаружения митохондриальных АФК (зеленый, Mito; красный, MitoSOX; синий, Hoechst).Масштабная линейка, 10 мкм. (BE) Средняя интенсивность флуоресценции (MFI) MitoSOX, определенная с помощью ImageJ (B; N = 5 в каждой группе; ** P <,01), и процент флуоресцентно-экспрессирующих MitoSOX, определенный с помощью анализа FACS (C) в TMAO- лечили БМДМ. Потенциал митохондриальной мембраны как отношение красного JC-1 к зеленому JC-1 определяли с помощью иммунофлуоресценции (D; масштабная шкала, 10 мкм), а дисфункциональное митохондриальное дыхание, помеченное MitoTracker Green , высокое MitoTracker Red , низкое , определяли с помощью FACS. анализ (E) в BMDM, обработанных 300 мкМ TMAO в течение 24 часов.(F) Репрезентативное иммунофлуоресцентное окрашивание NLRP3 (зеленый, Alexa Fluor 488), ASC (красный, Alexa Fluor 594), DAPI (синий) в BMDM, обработанных TMAO или TMAO + Tempo. Масштабная линейка, 5 мкм. (G) Вестерн-блоттинг анализ NLRP3, IL-1β и расщепленного IL-1β в BMDM, обработанных TMAO или TMAO + Tempo. (H) Секретируемый IL-1β в супернатантах, культивируемых BMDM, с TMAO, Tempo или TMAO+Tempo. N = 4 в каждой группе. ** P < .01. (I-J) Активность каспазы-1, определяемая как концентрация pNA (I; N = 4 в каждой группе; * P <.05, ** P <,01) или процент изотиоцианата флуоресцеина-FLICA (J) анализировали в BMDM, обработанных ТМАО или обработанных ТМАО + Tempo. (K) Экспрессию TNF-α (N = 3 в каждой группе) и IL-6 (N = 4 в каждой группе) определяли с помощью ELISA в супернатантах BMDM, культивируемых с TMAO, Tempo, TMAO+Tempo в течение 24 часов. ** P < .01. (L) Экспрессию CXCL9 и CXCL10 в BMDM определяли с помощью RT-PCR после обработки Tempo, TMAO или TMAO+Tempo в течение 24 часов. N = 3 в каждой группе. ** P < .01.
Интересно, что хотя TMAO-индуцированная поляризация NLRP3 блокировалась Mito-Tempo, продукция NLRP3 не ингибировалась (рис. 6F-G). Учитывая, что транскрипция NLRP3 может быть индуцирована активацией NF-κB, 32 мы дополнительно определили статус экспрессии NF-κB в BMDM при лечении ТМАО. Иммунофлуоресцентная визуализация показала, что NF-κB, индуцированный стимуляцией ТМАО, перемещался в ядра BMDM (дополнительная фигура 7A).Кроме того, фармакологический ингибитор NF-κB, триптолид, успешно ослаблял индуцированную ТМАО активацию p-NF-κB по мере снижения интенсивности флуоресценции p-NF-κB (дополнительная фигура 7B-C), отменял перинуклеарную агрегацию NLRP3 и подавлял NLRP3. производство (дополнительная фигура 7D-E), все из которых были индуцированы в BMDM, стимулированных ТМАО. Эти данные свидетельствуют о том, что ТМАО активирует NLRP3, способствуя ядерной транслокации NF-κB.
РТПХ является серьезным осложнением, возникающим в результате активации аллореактивных Т-клеток, что приводит к безрецидивной смертности при алло-ТГСК. 33 При РТПХ желудочно-кишечного тракта нарушенный слизистый барьер инициирует активацию антигенпрезентирующих клеток хозяина и дифференцировку донорских Т-клеток, что приводит к летальному усилению процесса патогенеза. 34 Доказана связь дисбактериоза гемостаза кишечной микробиоты с усилением РТПХ. 35,36 Все больше данных указывает на то, что живая микробная терапия, включая трансплантацию фекальной микробиоты и пробиотики, может восстановить микробное разнообразие и уменьшить тяжесть РТПХ. 37 Однако учет риска передачи фекальной микробиоты лекарственно-устойчивой бактериемии и неопределенность основного механизма ограничивают их трансляционное применение. 38 Необходимо изучить более целенаправленную терапию, касающуюся взаимосвязи между микробиотой и РТПХ. 39
Микробные метаболиты, репертуар, продуцируемый кишечными микроорганизмами, в основном включают SCFAs, 40 TMAO, 40 липополисахарид, 41 уремические токсины, 7 и желчные кислоты. 42 Накопленные данные показали, что микробные метаболиты кишечника играют важную роль в здоровье и заболеваниях человека. 43,44 Пищевые привычки изменяют определенное качество и количество кишечной микробиоты, что, с другой стороны, может влиять на микробные метаболиты и влиять на тяжесть РТПХ. 1,45 Было показано, что SCFAs и индол облегчают РТПХ, модулируя повреждение эпителиальных клеток кишечника 12 или через сигнальный путь интерферона типа I, 13 соответственно.Однако остается неясным, оказывают ли все микробные метаболиты кишечника сходную функцию при РТПХ. В этом исследовании мы сосредоточились на другом микробном метаболите, метаболизируемом холином ТМАО, чтобы прояснить его механизм регулирования РТПХ.
Повышенный уровень ТМАО в сыворотке коррелирует с воспалительными заболеваниями, такими как коронарный атеросклероз, 46 сахарный диабет 2 типа, 47 и хроническое заболевание почек. 48 В нашем исследовании лечение как холином, так и ТМАО увеличивало показатели, связанные с РТПХ, тогда как структурный аналог холина, ДМБ, обращал вспять прогрессирование РТПХ, вызванное холиновой диетой. Однако мы не обнаружили изменений в разнообразии кишечных бактерий и микробиоте, связанных с тяжестью РТПХ, что указывает на то, что РТПХ, вызванная диетой с высоким содержанием холина, не связана с микробным дисбиозом кишечника, но связана с его метаболитом ТМАО.
Интересно, что хотя у мышей с РТПХ было продемонстрировано, что CD4 + Т-клетки дифференцируются в связанные с РТПХ линии Th2 и Th27, а не линию Th3, 49 ни пролиферация Т-клеток, ни цитокины, высвобождаемые Т-клетками, не увеличивались при Стимуляция ТМАО в системе культивирования in vitro повышает вероятность того, что результат активации Th2 и Th27 у мышей, получавших ТМАО, не был прямым результатом контакта ТМАО с Т-клетками, а был следствием серийных иммунных ответов.Думая о потребности в антиген-презентации при РТПХ, опосредованной Т-клетками, мы, таким образом, сосредоточились на роли активации макрофагов в тяжести РТПХ, усиленной ТМАО.
Макрофаги обеспечивают микроокружение, в котором аллоантигены представляются донорским наивным CD4 + Т-клеткам, которые впоследствии дифференцируются в субпопуляции Th-клеток, экспрессирующих различные транскрипционные факторы и цитокины. 50 Создает среду для мощного ответа Th2 и Th27 макрофагами M1 при возникновении воспаления. 27,51 У мышей с РТПХ, получавших ТМАО, было увеличено процентное содержание макрофагов M1. Между тем, отличительная черта поляризованных цитокинов M1, полученных из BMDM, была повышена после лечения TMAO. Эти данные показали, что TMAO может контролировать поляризацию макрофагов M1, которая, в свою очередь, опосредует ответ активации Th2 и Th27.
Критическим этапом в поляризации M1 является активация воспаления NLRP3, требующая как праймирующей передачи сигналов (сигнал 1), так и активации передачи сигналов (сигнал 2). 29 В нашем исследовании было обнаружено, что ТМАО индуцирует транслокацию NF-κB и активирует инфламмасому NLRP3, делеция которой в BMDM приводит к затруднению индуцированной ТМАО поляризации M1. Эти результаты совпадают с предыдущим открытием в эндотелиальных клетках, что ТМАО может активировать инфламмасомы NLRP3 путем образования эндогенных митохондриальных АФК. 16,52
Активирующий сигнал, стимулируемый аденозинтрифосфатом, притоком кальция и продукцией АФК, приводит к расщеплению каспазы-1 и секреции провоспалительного цитокина IL-1β. 53 Было показано, что митохондриальные АФК повышены в макрофагах при обработке ТМАО, что было связано со снижением потенциала митохондриальной мембраны и нарушением митохондриального дыхания. Наши результаты были аналогичны предыдущему отчету о том, что ТМАО способствовал активации аортальных макрофагов и повышал экспрессию гена IL-1β в атеросклеротической бляшке. 54 Еще более примечательно то, что ТМАО сокращает выживаемость животных дикого типа, но не Nlrp3 -/- животных.Поляризация M1 в селезенке осталась неизменной у получавших аллогенный ТМАО Nlrp3 -/- реципиентов. Поскольку передача сигналов NLRP3 играет решающую роль в полном проявлении РТПХ, 55 наши результаты подтвердили эту функцию, показав, что общая выживаемость Nlrp3 -/- реципиентов не влияет на присутствие ТМАО.
Таким образом, РТПХ представляет собой длительный патологический процесс, при котором изменение диеты может изменить метаболизм микробиома и, таким образом, избежать летального исхода.Определение продуктов метаболизма кишечного микробиома, которые стимулируют или ингибируют РТПХ, поможет модифицировать трансляционную таргетную терапию для этого заболевания. Мы продемонстрировали, что ТМАО, генерируемый холином, как стимулятор прогрессирования РТПХ приводит к поляризации макрофагов M1 и, таким образом, активирует реакцию Th2 и Th27 (визуальное изображение). Наше исследование выявило недостающие звенья между холиновой диетой хозяина, микробными метаболитами и тяжестью РТПХ, раскрывая потенциал для облегчения РТПХ путем контроля потребления холина.Сочетание блокирования пути ТМАО с другими стратегиями, включая защиту SCFAs, поможет установить микроэкологический баланс и потенциально может стать целевым путем для контроля РТПХ и других воспалительных заболеваний.
За исходными данными обращайтесь к соответствующему автору.
Электронная версия этой статьи содержит дополнение данных.
Затраты на публикацию этой статьи были частично покрыты за счет оплаты страницы. Поэтому и исключительно для того, чтобы указать на этот факт, эта статья настоящим помечена как «реклама» в соответствии с разделом 1734 18 USC.
Авторы выражают особую благодарность Naiqing Zhao (Школа общественного здравоохранения Университета Фудань) за любезную помощь в статистическом анализе.
Эта работа была поддержана Национальным фондом естественных наук Китая (81870081, 109), Шанхайской программой выдающихся медицинских академических лидеров (2019LJ05), Шанхайской программой совместных инноваций в области регенеративной медицины и исследований стволовых клеток (2019CXJQ01), и Азиатско-американский партнерский фонд Milstein Medical (TC).
Добавил: Т.C. и HJ определили тему исследования, руководили исследовательскими экспериментами и написали статью; К.В. провел эксперименты, проанализировал данные и написал статью; Ю.Ю. внес вклад в дизайн исследования; H.Y., X.D., S.W., Z.S., F.W., H.F. и Q.L. помогли оптимизировать условия эксперимента.
Раскрытие информации о конфликте интересов: авторы заявляют об отсутствии конкурирующих финансовых интересов.
Адрес для переписки: Тонг Чен, отделение гематологии, больница Хуашань, Университет Фудань, 12 Улумуци Мидл Роад, Шанхай 200040, Китай; электронная почта: chentong@fudan.образование.сп; и Хуа Цзян, отделение гинекологии, больница акушерства и гинекологии, Университет Фудань, 419 Fangxie Rd, Шанхай 200011, Китай; электронная почта: [email protected].
.